UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K
| |
☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2022
or
| |
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number: 0-19961

ORTHOFIX MEDICAL INC.
(Exact name of registrant as specified in its charter)
| | |
Delaware | | 98-1340767 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification No.) |
| |
3451 Plano Parkway, Lewisville, Texas | | 75056 |
(Address of principal executive offices) | | (Zip Code) |
(214) 937-2000
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | | | |
Common Stock, $0.10 par value | | OFIX | | Nasdaq Global Select Market |
(Title of Class) | | (Trading Symbol) | | (Name of Exchange on Which Registered) |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | |
Large accelerated filer | | ☐ | | Accelerated filer | | ☒ | | Emerging Growth Company | | ☐ |
Non-accelerated filer | | ☐ | | Smaller reporting company | | ☐ | | | | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. §7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The aggregate market value of registrant’s common stock held by non-affiliates, based upon the closing price of the common stock on the last business day of the fiscal quarter ended June 30, 2022, as reported by the Nasdaq Global Select Market, was approximately $470.8 million.
As of March 1, 2023, 36,455,564 shares of common stock were issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Certain sections of the registrant’s definitive proxy statement to be filed with the Commission in connection with the Orthofix Medical Inc. 2022 Annual Meeting of Shareholders are incorporated by reference in Part III of this Annual Report.
Orthofix Medical Inc.
Form 10-K for the Year Ended December 31, 2022
Table of Contents
Forward-Looking Statements
This Annual Report contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended (“the Exchange Act”), and Section 27A of the Securities Act of 1933, as amended, relating to our business and financial outlook, which are based on our current beliefs, assumptions, expectations, estimates, forecasts, and projections. In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “should,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “projects,” “intends,” “predicts,” “potential,” or “continue” or other comparable terminology. Forward-looking statements include, but are not limited to, statements about:
•our intentions, beliefs, and expectations regarding our operations, sales, expenses, and future financial performance;
•our intentions, beliefs, and expectations regarding the anticipated benefits of the recent merger with SeaSpine Holdings Corporation, including the anticipated synergies and cost-savings from the merger;
•our plans for future products and enhancements of existing products;
•anticipated growth and trends in our business;
•the timing of and our ability to maintain and obtain regulatory clearances or approvals;
•our belief that our cash and cash equivalents, investments, and access to our revolving line of credit will be sufficient to satisfy our anticipated cash requirements;
•our expectations regarding our revenues, customers, and distributors;
•our expectations regarding our costs, suppliers, and manufacturing abilities;
•our beliefs and expectations regarding our market penetration and expansion efforts;
•our expectations regarding the benefits and integration of acquired businesses and/or products (including in connection with our merger with SeaSpine Holdings Corporation in January 2023) and our ability to make future acquisitions and successfully integrate any such future-acquired businesses;
•our anticipated trends and challenges in the markets in which we operate; and
•our expectations and beliefs regarding and the impact of investigations, claims and litigation.
These forward-looking statements are not guarantees of future performance and involve risks, uncertainties, estimates, and assumptions that are difficult to predict. Any or all forward-looking statements that we make may turn out to be wrong (due to inaccurate assumptions that we make or otherwise) and our actual outcomes and results may differ materially from those expressed in these forward-looking statements. Potential risks and uncertainties that could cause actual results to differ materially include, but are not limited to, those set forth in Part I, Item 1A under the heading “Risk Factors”, Part II, Item 7 “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere throughout this Annual Report and in any other documents incorporated by reference to this Annual Report. You should not place undue reliance on any of these forward-looking statements. Further, any forward-looking statement speaks only as of the date hereof, unless it is specifically otherwise stated to be made as of a different date. We undertake no obligation to update, and expressly disclaim any duty to update, our forward-looking statements, whether as a result of circumstances or events that arise after the date hereof, new information, or otherwise.
Trademarks
Solely for convenience, our trademarks and trade names in this Annual Report are referred to without the ® and ™ symbols, but such references should not be construed as any indicator that we will not assert, to the fullest extent under applicable law, our rights thereto.
PART I
Item 1. Business
In this Annual Report, the terms “we,” “us,” “our,” “Orthofix,” and “the Company” refer to the combined operations of Orthofix Medical Inc. and its consolidated subsidiaries and affiliates, unless the context requires otherwise.
Company Overview
Following our recent merger with SeaSpine Holdings Corporation ("SeaSpine"), the newly merged Orthofix-SeaSpine organization is a leading global spine and orthopedics company with a comprehensive portfolio of biologics, innovative spinal hardware, bone growth therapies, specialized orthopedic solutions and a leading surgical navigation system. Our products are distributed in approximately 68 countries worldwide through a combination of direct and indirect sales representatives and stocking distributors.
We are headquartered in Lewisville, Texas and have primary offices in Carlsbad, CA, with a focus on spine and biologics product innovation and surgeon education, and Verona, Italy, with an emphasis on product innovation, production, and medical education for orthopedics. Our combined global R&D, commercial and manufacturing footprint also includes facilities and offices in Irvine, CA, Toronto, Canada, Sunnyvale, CA, Wayne, PA, Olive Branch, MS, Maidenhead, UK, Munich, Germany, Paris, France and Sao Paulo, Brazil.
The Company originally was formed in 1987 in Curaçao as “Orthofix International N.V.” In 2018, we completed a change in our jurisdiction of organization from Curaçao to the State of Delaware (the “Domestication”) and changed our name to “Orthofix Medical Inc.” As a result, we are a corporation existing under the laws of the State of Delaware.
Our merger with SeaSpine was completed on January 5, 2023, with SeaSpine continuing as a wholly-owned subsidiary of Orthofix following the transaction. Orthofix, as the corporate parent entity in the combined company structure, continues to trade on NASDAQ under the symbol “OFIX.” The parent company will be renamed at a later date, and until then, will continue to be known as Orthofix Medical Inc. The disclosures in this Item 1 of this Annual Report under the heading “Business” speak to the combined company subsequent to the merger unless otherwise noted. However, the financial results described herein relate, except as otherwise expressly noted herein, to Orthofix on a standalone basis without to giving effect to merger and, accordingly, do not include the results of SeaSpine. Future filings, beginning with our Quarterly Report on Form 10-Q for the fiscal quarter ending March 31, 2023, will reflect the results of the combined Orthofix-SeaSpine organization.
Available Information and Orthofix Website
Our filings with the Securities and Exchange Commission (“SEC”), including our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, Proxy Statements for Meetings of Shareholders, any registration statements, and amendments to those reports, are available free of charge on our website as soon as reasonably practicable after they are filed with, or furnished to, the SEC. Information contained in our website or connected to our website is not incorporated by reference into this Annual Report. Our website is located at www.orthofix.com. Our SEC filings are also available on the SEC website at www.sec.gov.
Business Segments
Historically, Orthofix has managed the business by two reporting segments, Global Spine and Global Orthopedics, which account for 77% and 23%, respectively, of Orthofix's total net sales in 2022. The chart below presents Orthofix's net sales, which includes product sales and marketing service fees, by reporting segment for each of the years ended December 31, 2022, 2021, and 2020. As noted above, these amounts do not include the net sales of SeaSpine.
4
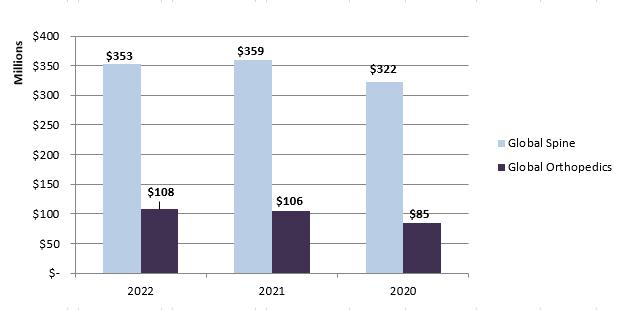
SeaSpine has historically managed its business as one operating segment, but with revenue reported in two product categories: (i) Biologics (formerly recognized as Orthobiologics) and (ii) Spinal Implants and Enabling Technologies. For purposes of this Annual Report, SeaSpine's historical business description is included within this discussion of Business Segments as a separate segment. Following the merger with SeaSpine, which was completed on January 5, 2023, we expect to reassess our reporting segments in the first quarter of 2023 based on how the operations of the newly combined company will be managed.
Financial information regarding our reportable business segments and certain geographic information is included in Part II, Item 7 of this Annual Report under the heading “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and Note 16 of the Notes to the Consolidated Financial Statements in Item 8 of this Annual Report.
Global Spine
Within the Global Spine segment, we provide implantable medical devices, biologics, and other regenerative solutions which aim to restore the quality of life of patients suffering from diseases and traumas of the spine. We offer a variety of treatment solutions that uniquely incorporate multiple treatment modalities, such as mechanical, biological, and electromagnetic modes, to achieve desired clinical outcomes.
Global Spine Strategy
Our strategy for the Global Spine segment is to drive business growth through organic and inorganic innovation, physician collaboration, and partnerships with dedicated and high-performing commercial sales channels. Growth initiatives include:
•Continued expansion of our presence in the U.S cervical disc replacement market through surgeon training, publication of clinical evidence to include long-term real world evidence, patient education, and sales channel support
•A regular cadence of new product launches supporting our spine implant, biologics, and bone growth therapies portfolios
•Ongoing, global sales channel optimization
•Reinforcement of our bone growth stimulation business through the collection and dissemination of clinical evidence, and the delivery of new and novel value-added services
•Conducting clinical research to support and broaden our spine implant, biologics, and bone growth stimulation portfolios
•Acquiring or licensing products, technologies, and companies to further expand the spine portfolio
•Attracting, developing, and retaining key talent
5
Global Spine Principal Products
The Global Spine reporting segment is largely represented by three principal product categories, i) Bone Growth Therapies, ii) Spinal Implants, and iii) Biologics. Each of these product categories are further described below:
Bone Growth Therapies
Within the Bone Growth Therapies product category, we manufacture, distribute, and provide support services for market-leading bone growth stimulation devices that enhance bone fusion. These class III medical devices are indicated as an adjunctive, noninvasive treatment to improve fusion success rates in the cervical and lumbar spine as well as a therapeutic treatment for non-spinal, appendicular fractures that have not healed ("nonunions"). Several devices in our portfolio utilize our patented pulsed electromagnetic field (“PEMF”) technology, the safety and efficacy of which is supported by basic mechanism of action data in the scientific literature, as well as published data from level one randomized controlled clinical trials. A new addition to our stimulation portfolio utilizes our low intensity pulsed ultrasound ("LIPUS"), a technology also supported by strong basic science and published clinical literature. Orthofix is the only manufacturer which offers both PEMF and LIPUS technologies. We sell these products almost exclusively in the U.S. using distributors and direct sales representatives to provide our devices to healthcare providers and their patients.
Spinal Implants
Within the Spinal Implants product category, we design, develop, and market a portfolio of motion preservation and fixation implant products used in surgical procedures of the spine. We distribute these products globally through a network of distributors and sales representatives to sell spine products to facilities that conduct spine care, including hospitals, ambulatory surgery centers, and out-patient hospitals.
Biologics
Within the Biologics product category, we offer a portfolio of products and tissue forms that allow physicians to successfully treat a variety of spinal and orthopedic conditions. We market tissue forms provided by MTF Biologics (“MTF”) to spine care facilities and surgeons, primarily in the U.S., through a network of independent distributors and sales representatives. Our partnership with MTF allows us to exclusively market the Virtuos Lyograph, Trinity ELITE, FiberFuse Advanced, FiberFuse Strip, and certain other tissue forms for musculoskeletal defects to enhance bony fusion. In addition, we market regenerative non-tissue biologic solutions derived from synthetic materials. Opus BA, and Opus MG Set represent our current synthetic, biologic offering.
The following table and discussion identify our principal Global Spine products by trade name and describe their primary applications:
| | |
Product | | Primary Application |
| |
Bone Growth Therapies Products | | |
| | |
CervicalStim Spinal Fusion Therapy | | PEMF non-invasive cervical spinal fusion therapy used to enhance bone growth |
| |
SpinalStim Spinal Fusion Therapy | | PEMF non-invasive lumbar spinal fusion therapy used to enhance bone growth |
| |
PhysioStim Bone Healing Therapy | | PEMF non-invasive appendicular skeleton healing therapy used to enhance bone growth in nonunion fractures |
| | |
AccelStim | | LIPUS healing therapy used to enhance bone growth in certain fresh, distal radius and tibial diaphysis fractures |
| | |
Spinal Implants Products | | |
| | |
M6-C Artificial Cervical Disc | | A next-generation artificial disc developed to replace an intervertebral disc damaged by cervical disc degeneration; the only artificial cervical disc that mimics the anatomic structure of a natural disc by incorporating an artificial viscoelastic nucleus and fiber annulus into its design |
| | |
M6-L Artificial Lumbar Disc | | A next-generation artificial disc developed to replace an intervertebral disc damaged by lumbar disc degeneration; the only artificial lumbar disc that |
6
| | |
| | mimics the anatomic structure of a natural disc by incorporating an artificial viscoelastic nucleus and fiber annulus into its design (Not available in the U.S.) |
| | |
FIREBIRD / FIREBIRD NXG Spinal Fixation System | | A system of rods, crossbars, and modular pedicle screws designed to be implanted during a posterior lumbar spine fusion procedure |
| | |
FORZA XP Expandable Spacer System | | A titanium expandable spacer system for posterior lumbar interbody fusion (“PLIF”) and transforaminal lumbar interbody fusion (“TLIF”) procedures featuring a large graft window with the ability to pack post expansion in situ |
| | |
FORZA PEEK / Titanium Composite (“PTC”) Spacer System | | A posterior lumbar interbody with 3D printed porous titanium end plates that may promote bone ingrowth and a polyetheretherketones (“PEEK”) core to maintain imaging characteristics |
| | |
FORZA Spacer System | | PEEK interbody devices for PLIF and TLIF procedures |
| | |
FORZA Ti Spacer System | | Fully 3D printed titanium devices for PLIF and TLIF procedures |
| | |
CENTURION Posterior Occipital Cervico-Thoracic (“POCT”) System | | A multiple component system comprised of a variety of non-sterile, single use components made of titanium alloy or cobalt chrome that allow the surgeon to build a spinal implant construct |
| |
PHOENIX Minimally Invasive Spinal Fixation System | | A multi-axial extended reduction screw body used with the Firebird Spinal Fixation System designed to be implanted during a posterior thoracolumbar spine fusion procedure |
| | |
CONSTRUX Mini PTC Spacer System | | An anterior cervical interbody with 3D printed porous titanium end plates that may promote bone ingrowth and a PEEK core to maintain imaging characteristics |
| | |
CONSTRUX Mini Ti Spacer System | | Fully 3D printed titanium anterior cervical interbody spacer system |
| |
CETRA Anterior Cervical Plate System | | An anterior cervical plate system offering a low profile plate with an intuitive locking mechanism, large graft windows, a high degree of screw angulation, and simplified instrumentation |
| |
JANUS Midline Fixation Screw | | An addition to the Firebird Spinal Fixation System designed to achieve more cortical bone purchase in the medial to lateral trajectory, when compared to traditional pedicle screws, and that provides surgeons with the option of a midline approach |
| | |
LONESTAR Cervical Stand Alone | | A stand-alone spacer system designed to provide the biomechanical strength to a traditional or minimal invasive anterior cervical discectomy and fusion procedure with less disruption of patient anatomy and to preserve the anatomical profile |
| |
PILLAR SA PTC PEEK Spacer System | | A standalone anterior lumbar interbody fusion ("ALIF") interbody with 3D printed porous titanium end plates that may promote bone ingrowth and a PEEK core to maintain imaging characteristics |
| | |
SKYHAWK Lateral Interbody Fusion System & Lateral Plate System | | Provides a complete solution for the surgeon to perform a lateral lumbar interbody fusion, an approach to spinal fusion in which the surgeon accesses the intervertebral disc space using a surgical approach from the patient’s side that disturbs fewer structures and tissues |
| | |
FIREBIRD SI | | A minimally invasive screw system that is intended for fixation of sacroiliac joint disruptions in skeletally mature patients |
| | |
7
| | |
Biologics Technologies | | |
| | |
Virtuos Lyograft | | A first-of-its-kind, shelf-stable and complete autograft substitute for spine and orthopedic procedures provided in a room-temperature, ready-to-use, moldable form |
| |
Trinity ELITE | | A fully moldable allograft with viable cells used during surgery that is designed to aid in the success of a spinal fusion or bone fusion procedure |
| | |
FiberFuse Advanced | | An allograft comprised of a mixture of cancellous bone and demineralized cortical bone fibers that creates a natural scaffold for revascularization, cellular ingrowth, and new bone formation |
| | |
FiberFuse Strip | | A preformed allograft that consists of mineralized cancellous bone and demineralized cortical fibers, providing an ideal matrix for bone healing |
| | |
O-Genesis Graft Delivery | | A bone graft delivery system, which is provided in a sterile, single-use form |
| |
Opus Mg Set | | An injectable, moldable, and biocompatible bone void filler that will harden in-situ at the defect site |
| | |
Opus BA | | A synthetic osteoconductive scaffold that is compression resistant, fully resorbable, and easily customizable for a range of clinical applications |
| | |
Legacy Demineralized Bone Matrix ("DBM") | | A ready-to-use, flowable DBM putty |
Bone Growth Therapies — Spinal Therapy
Our bone growth therapy devices used in spinal applications are designed to enhance bone growth and improve the success rate of certain spinal fusion procedures by stimulating the body’s own natural healing mechanism post-surgically. These non-invasive portable devices are intended to be used as part of a home treatment program prescribed by a physician.
We offer two spinal fusion therapy devices: the SpinalStim and CervicalStim devices. Our stimulation products use a PEMF technology designed to enhance the growth of bone tissue following surgery and are placed externally over the site to be healed. Research data shows that our PEMF signal induces mineralization and results in a process that stimulates new regeneration at the spinal fusion site. Some spine fusion patients are at greater risk of not achieving a solid fusion of new bone around the fusion site. These patients typically have one or more risk factors, such as smoking, obesity, or diabetes, or their surgery involves the revision of a failed fusion or the fusion of multiple levels of vertebrae in one procedure. For these patients, post-surgical bone growth therapy has been shown to significantly increase the probability of fusion success.
The SpinalStim device is a non-invasive spinal fusion stimulator system designed for the treatment of the lumbar region of the spine. The device uses proprietary technology and a wavelength to generate a PEMF signal. The U.S. Food and Drug Administration (the “FDA”) has approved the SpinalStim system as a spinal fusion adjunct to increase the probability of fusion success and as a non-operative treatment for salvage of failed spinal fusion at least nine months post-operatively.
Our CervicalStim product remains the only FDA-approved bone growth stimulator on the market indicated for use as an adjunct to cervical spine fusion surgery. It is indicated for patients at high-risk for non-fusion.
The SpinalStim and CervicalStim systems are accompanied by an application for mobile devices called STIM onTrack. The mobile app includes a first-to-market feature that enables physicians to remotely view patient adherence to prescribed treatment protocols and patient reported outcome measures. Designed for use with smartphones and other mobile devices, the STIM onTrack tool helps patients follow their prescription with daily treatment reminders and a device usage calendar. The app is free and available through the Android and Apple App Stores.
Bone Growth Therapies — Orthopedic Therapy
Our PhysioStim bone healing therapy products use PEMF technology similar to that used in our spine stimulators. The primary difference is that the PhysioStim devices are designed for use on the appendicular skeleton.
A bone’s regenerative power results in most fractures healing naturally within a few months. However, in the presence of certain risk factors, some fractures do not heal or heal slowly, resulting in “nonunions.” Traditionally, orthopedists have treated such nonunion conditions surgically, often by means of a bone graft with fracture fixation devices, such as bone plates, screws, or
8
intramedullary rods. These are examples of “invasive” treatments. Our patented PhysioStim bone healing therapy products are designed to use a low level of PEMF signals to noninvasively activate the body’s natural healing process. The devices are anatomically designed, allowing ease of placement, patient mobility, and the ability to cover a large treatment area.
Similar to our SpinalStim and CervicalStim systems, the PhysioStim device is also accompanied by the STIM onTrack mobile app, enabling physicians treating patients with nonunion fractures to remotely view and assess patient adherence to prescribed treatment protocols and patient reported outcome measures.
The AccelStim device provides a safe and effective nonsurgical treatment to improve nonunion fracture healing and accelerate the healing of indicated fresh fractures. The device stimulates the bone’s natural healing process through LIPUS waves to the fracture site.
Spinal Implants — Motion Preservation Solutions
Our M6-C cervical and M6-L lumbar artificial discs are used to treat patients suffering from degenerative disc disease of the spine. The M6 discs are the only FDA-approved artificial discs that mimic the anatomic structure of a natural disc by incorporating an artificial viscoelastic nucleus and fiber annulus into their design. Like a natural disc, this unique construct allows for shock absorption at the implanted level, as well as provides a controlled range of motion when the spine transitions in its combined complex movements. Both discs have European Commission CE mark approval and in February 2019, we received FDA approval of the M6-C artificial cervical disc to treat patients with a single-level cervical disc degeneration. We released the M6-C artificial cervical disc in the U.S. in 2019 through a controlled market launch accompanied by an extensive training and education curriculum for surgeons. The M6-C disc has become our leading spinal implant device and has contributed significantly to our growth in recent years. In addition, we have initiated a U.S. 2-level investigational device exemption (“IDE”) study for the M6-C artificial cervical disc, which is currently enrolling.
Spinal Implants — Spinal Fixation Solutions
We provide a wide array of implants designed for use primarily in cervical, thoracic, and lumbar fusion surgeries. These implants are made of either metal or a thermoplastic compound called PEEK. The majority of the implants that we offer are made of titanium metal. The Firebird Spinal Fixation System, the Phoenix Minimally Invasive Spinal Fixation System, and the Centurion POCT Systems are sets of rods, cross connectors, and screws that are implanted during posterior fusion procedures. The Firebird Modular and pre-assembled Spinal Fixation Systems are designed to be used in either open or minimally-invasive posterior lumbar fusion procedures. To complement our plates, rods, and screw fixation options, we offer an entire portfolio of cervical and thoracolumbar Titanium and PEEK interbody devices within our Pillar and Forza product lines. We have recently introduced two, new 3D printed interbody solutions, Construx Mini Ti for cervical and Forza Ti for posterior lumbar implantation. This interbody portfolio includes two stand-alone devices, Lonestar and Pillar SA, as well as the Construx Mini PTC system, a novel titanium composite spacer, which offers a superior alternative to other plasma spray coated options currently available on the market. We also offer specialty plates and screws that are used in less common procedures.
Biologics — Regenerative Solutions
The premium biologics tissues we market include the Virtuos Lyograft and Trinity ELITE tissue forms, which are cortico-cancellous allografts that retain the inherent growth factors and viable cells found in bone. They are used during surgery in the treatment of musculoskeletal defects for bone reconstruction and repair. These allografts are intended to offer a viable alternative to an autograft procedure, as harvesting autograft has been shown to add risk of an additional surgical procedure and related patient discomfort in conjunction with a repair surgery. Virtuos Lyograft is particularly unique in that it is a first-of-its-kind, shelf-stable and complete autograft substitute for spine and orthopedic procedures provided in a room-temperature, ready-to-use, moldable form.
The FiberFuse Advanced tissue is a tissue form with handling characteristics analogous to the Trinity ELITE product without compromising bone content. It provides an advanced demineralized bone offering that leverages fiber technology with the advantages of ingrowth that cancellous bone provides and expands the offering to address a broader scope of surgical applications. FiberFuse Strip is a preformed allograft form of FiberFuse Advanced that consists of mineralized cancellous bone and demineralized cortical fibers, providing an ideal matrix for bone healing. Legacy DBM is a ready-to-use, flowable, demineralized bone putty and provides a cost-effective option without compromising clinical experience.
We receive marketing fees through our collaboration with MTF for the Virtuos, Trinity ELITE, FiberFuse Advanced, FiberFuse Strip, Legacy DBM and certain other tissues. MTF processes the tissues, maintains inventory, and invoices hospitals, surgery centers, and other points of care for service fees, which are submitted by customers via purchase orders. We have exclusive worldwide rights to market the Virtuos and Trinity ELITE and exclusive rights to market the FiberFuse Advanced and FiberFuse Strip tissues in the U.S.
9
Regarding synthetic, biologic solutions, we offer Opus BA and Opus Mg Set. Opus BA is a synthetic bioactive solution that is easily hydrated and flexible. A carefully selected trifecta of components creates an ideal environment for bone growth building on the earlier generations of synthetic bone grafts. Opus Mg Set is an injectable, moldable, and biocompatible bone void filler that will harden in-situ at the defect site.
To date, our Biologics products are offered primarily in the U.S. market due in part to restrictions on providing U.S. human donor tissue in other countries.
Global Spine Future Product Applications
We remain very active with multiple internal developments to support future, new technology commercialization efforts. These new technologies will apply to both the cervical and thoracolumbar spinal anatomy. In addition, we remain active in evaluating external licensing and acquisition opportunities to add implant, biologics, and other emerging technologies to our spine portfolio. We expect that the contribution of new, internally developed technologies and undefined external acquisitions will be the primary driver of future growth.
Regarding our Bone Growth Therapy business, we have participated in research at the Wake Forest University Health Sciences, Chinese University of Hong Kong, and University of California San Francisco, where scientists conducted animal and cellular studies to identify the mechanisms of action of our PEMF signals on bone, cartilage, meniscus, nerve, and efficacy of healing. From these efforts, some studies have been published in peer-reviewed journals. Among other insights, the studies illustrate positive effects of PEMF on callus formation and bone strength, meniscus and nerve injury repair, as well as proliferation and differentiation of cells involved in tissue regeneration and healing. Furthermore, we believe that the previous research work with Cleveland Clinic, the Chinese University of Hong Kong, and the University of Pennsylvania, allowing for characterization and demonstration of the Orthofix new PEMF waveform, is paving the way for signal optimization for a variety of new applications and indications. This collection of pre-clinical data, along with additional clinical data, could represent new clinical indication opportunities for our regenerative stimulation solutions. In addition, we also have initiated a U.S. 2-Level IDE study for the M6-C artificial cervical disc.
Global Orthopedics
The Global Orthopedics reporting segment offers products and solutions for limb deformity correction and complex limb reconstruction with a focus on use in trauma, pediatrics, and foot and ankle procedures. This reporting segment specializes in the design, development, and marketing of external and internal fixation orthopedic products that are coupled with enabling digital technologies to serve the complete patient treatment pathway. We sell these products through a global network of distributors and sales representatives to hospitals, healthcare organizations, and healthcare providers.
Global Orthopedics Strategy
Our strategy for the Global Orthopedics reporting segment is to continue to offer pioneering limb reconstruction and deformity correction procedural solutions that address the entire patient treatment pathway.
Our key strategies in this segment are:
•Expand our position as the worldwide leader in complex deformity and limb reconstruction, including both internal and external solutions, through a patient-centric approach and digital treatment journey
•Promote the advantages of our expansive pediatric product portfolio and support tools
•Leverage our cross-product OrthoNext digital platform, a uniquely developed pre and post planning digital platform, that allows our clinicians to pre-plan surgery for patients so they can start surgeries with a greater degree of confidence, reduce surgical times, enable better outcomes and follow up post operatively to evaluate their chosen surgical plan success
•Expand our foot and ankle portfolio by building on our historical position as a company highly focused on addressing complex and challenging conditions and remaining at the forefront of innovation in helping surgeons and patients alike in the management of Charcot foot and ankle
•Promote and invest in our Fitbone intermedullary limb lengthening platform, which together with our external fixation products, offers surgeons internal and external solutions for limb lengthening and deformity correction
•Within the orthopedic trauma segment, continue to focus on open and complex fracture management
•Collaborate with physicians and healthcare partners to improve patients’ lives through technology, digital transformation, clinical evidence, and our industry-leading medical education programs, such as Orthofix Academy
10
•Continue the strong pace of new product launches
•Acquire or license products, technologies, and companies to support these market opportunities.
Global Orthopedics Focus Products
Global Orthopedics offers a comprehensive line of limb reconstruction and complex deformity correction technologies. We provide innovative and minimally invasive extremity solutions to help surgeons improve their patients' quality of life, which are designed to address the lifelong bone and joint health needs of patients of all ages. In addition, our well-rounded product lines offer internal and external fixation solutions for pediatrics, limb reconstruction, trauma, and foot and ankle specialties.
Our fracture repair solutions comprise a wide range of devices designed for specific anatomical areas. The philosophy underlying these devices is to provide adequate stability and to allow for early functional recovery, thereby improving patients’ quality of life. Our goal is to offer devices that enable a simple, standardized approach for reproducible results.
Our trauma products consist of a comprehensive portfolio of ready-to-use, sterile, dedicated implant kits designed for a wide range of anatomical sites.
The following table and discussion identifies the principal Global Orthopedics products by trade name and describes their primary applications:
| | |
Product | | Primary Application |
| |
TrueLok | | A surgeon-designed, lightweight external fixation system for trauma, limb lengthening, and deformity correction, which consists of circular rings and semi-circular external supports centered on the patient’s limb and secured to the bone by crossed, tensioned wires, and half pins |
| | |
TrueLok Hexapod System (“TL-HEX”) | | A hexapod external fixation system for trauma and deformity correction with associated software, designed as a three-dimensional bone segment reposition module to augment the previously developed TrueLok frame. The system consists of circular and semi-circular external supports, secured to the bones by wires and half pins and interconnected by six struts, which allows multi-planar adjustment of the external supports. The rings’ positions are adjusted either rapidly or gradually in precise increments to perform bone segment repositioning in three-dimensional space |
| | |
TrueLok EVO | | A modular circular external fixation system that features both radiolucent rings and struts to enable clear radiographic visualization to allow physicians to better assess bone anatomy both during surgery and post-operative care |
| | |
FITBONE Intramedullary Limb-Lengthening System | | An intramedullary lengthening system intended for limb lengthening of the femur and tibia, surgically implanted in the bone through a minimally invasive procedure; it includes an external telemetry control set that manages the distraction process, and is the only intramedullary limb lengthening system with an FDA-cleared pediatric indication |
| | |
Pediatric Portfolio | | Our pediatric solutions include a range of products and resources dedicated to pediatrics and young adults with bone fractures and deformities. With our 360° approach to the patient journey we provide dedicated tools to treat all stages of the healing process: collaterals, educational games, software applications, and patient apps for post-operative management Our pediatric solutions portfolio includes, among the others: - A complete line of nailing systems for trauma and limb reconstruction, including our elastic nail, MJ-FLEX, and our rigid intramedullary nail for adolescents, Agile Nail; - The Galaxy Fixation Pediatric System; |
11
| | |
| | - The eight-Plate Guided Growth System (“eight-Plate”) and the eight-Plate Guided Growth System+ (“eight-Plate Plus”); - The JuniOrtho Plating System |
| | |
Galaxy Fixation System | | A pin-to-bar system for temporary and definitive fracture fixation, in the upper and lower limbs. The system incorporates a streamlined combination of clamps, with both pin-to-bar and bar-to-bar coupling capabilities, offering a complete range of applications, including specific anatomic units for the shoulder, elbow and wrist. The latest version, Galaxy Gemini, includes a universal clamp and other updates to better streamline surgical procedures |
| | |
Galaxy Fixation Shoulder | | A unique solution for the treatment of proximal humeral fractures |
| | |
Ankle Hindfoot Nail (“AHN”) | | A differentiated solution for hindfoot fusions that includes a revision option to address more large bone defects and more complex hindfoot pathologies |
| | |
G-BEAM Fusion Beaming System | | A system designed to address the specific demands of advanced deformity and trauma reconstructions of foot and ankle applications, such as Charcot, requiring fusion of the medial and/or lateral columns, with or without corrective osteotomies as well as for joint fusions within the mid- and hindfoot |
| | |
OSCAR | | An ultrasonic powered surgical system for revision arthroplasty |
| | |
External Fixators | | External fixation, including our limb-lengthening systems, ProCallus, XCaliber, Pennig, Radiolucent Wrist Fixators, and Calcaneal Fixator |
| |
eight-Plate and eight-Plate Plus | | The first and still market-leading system for gradual correction of the growth plate in pediatric patients |
| |
LRS advanced Limb Reconstruction System | | An external fixation solution for limb lengthening and corrections of deformity, which uses callus distraction to lengthen bone in a variety of procedures, including monofocal lengthening and corrections of deformity; its multifocal procedures include bone transport, simultaneous compression and distraction at different sites, bifocal lengthening, and correction of deformities with shortening |
| | |
OrthoNext Digital Platform | | A digital platform software developed specifically for use with the JuniOrtho Plating System and Fitbone Intramedullary Limb Lengthening System, which enables the surgeon to accurately plan the deformity correction and osteotomy position as well as visualize the implant in relation to the anatomy |
We provide internal and external fixation solutions for extremity repair and deformity correction, both for adults and children. Our fracture repair products consist of fixation devices designed to stabilize a broken bone until it can heal. With these devices, we can treat simple and complex fracture patterns, along with achieving deformity corrections.
External Fixation
External fixation devices are used to stabilize fractures and offer an ideal treatment for complex fractures, fractures near the joints, and in patients with known risk factors or co-morbidities. The treatment is minimally invasive and allows external manipulation of the bone to obtain and maintain final bone alignment (reduction). The bone is fixed in this way until healing occurs. External fixation allows small degrees of micromotion (dynamization), which promotes blood flow at the fracture site, and accelerates the bone healing process. External fixation devices may also be used temporarily in complex trauma cases to stabilize the fracture prior to treating it definitively. In these situations, the device offers rapid fracture stabilization, which is important in life-saving as well as limb salvage procedures.
We offer most of our products in sterile packaging, which fulfills the need of a streamlined and ready-to-use set of products, particularly in trauma applications where timing is crucial.
Examples of our external fixation devices include the TrueLok, TL-HEX, TrueLok Evo, the Galaxy and Galaxy Gemini Fixation Systems, and the LRS Advanced Limb Reconstruction System.
12
Internal Fixation
Internal fixation devices consist of either long rods, commonly referred to as nails, or plates that are attached to the bone with the use of screws. Nails and plates come in various sizes, depending on the bone that requires treatment. A nail is inserted into the medullary canal of a fractured long bone of the human arm or leg (e.g., humerus, femur, or tibia). Alternatively, a plate is attached by screws to an area such as a broken wrist, hip, or foot. Examples of our internal fixation devices include Chimaera, AHN, and the G-BEAM Fusion Beaming System.
Acquired in March 2020, the FITBONE Intramedullary Limb Lengthening System provides an internal option for limb lengthening of the femur and tibia and provides Orthofix with the most complete limb reconstruction portfolio on the market. We are continuing to invest in the FITBONE technology platform in order to offer surgeons more solutions for deformity correction.
In addition to treating bone fractures, we also design, manufacture, and distribute devices intended to treat congenital bone conditions, such as angular deformities (e.g., bowed legs in children), degenerative diseases, and conditions resulting from a previous trauma. An example of a product offered in this area is the eight-Plate Plus Guided Growth System.
SeaSpine
SeaSpine's business focuses on the design, development, and commercialization of surgical solutions for the treatment of patients suffering from spinal disorders. We have a comprehensive portfolio of biologics and spinal implant solutions, as well as a surgical navigation system, to meet the varying combinations of products that neurosurgeons and orthopedic spine surgeons need to perform fusion procedures in the lumbar, thoracic, and cervical spine. We believe this broad combined portfolio is essential to meet the “complete solution” requirements of these surgeons.
SeaSpine has historically reported revenue in two product categories: (i) Biologics (formerly recognized as Orthobiologics) and (ii) Spinal Implants and Enabling Technologies. Our Biologics products consist of a broad range of advanced and traditional bone graft substitutes designed to improve bone fusion rates following a wide range of orthopedic surgeries, including spine, hip, and extremities procedures. Our Spinal Implants and Enabling Technologies portfolio consists of an extensive line of products and image-guided surgical solutions to facilitate spinal fusion in degenerative, minimally invasive surgery ("MIS"), and complex spinal deformity procedures. Expertise in biologic sciences and spinal implants, software and advanced optics product development allows SeaSpine to offer surgeon customers a differentiated portfolio and a complete solution to meet their patients' fusion requirements.
SeaSpine Strategy
Our goal in the SeaSpine business is to continue to scale our business in order to enhance our market position in biologics and become a leader in the spinal implant and image guided surgery market. To achieve our goal, we are investing in these strategies:
•Continue to increase our research and development activities to bring new products and techniques to market
•Continue to increase the quality, size, exclusivity, and geographic breadth of our network of independent sales agents in the U.S.
•Invest in the further development of our pre-clinical and clinical programs designed to generate peer-reviewed scientific evidence in support of our products
•Continue to pursue strategic alliances and acquisition opportunities to enhance our product offerings
SeaSpine Principal Products
SeaSpine is largely represented by two principal product categories, i) Biologics and ii) Spinal Implants and Enabling Technologies. Each of these product categories are further described below:
13
Biologics
Our Biologics products are used in orthopedic and dental procedures and consist of a broad range of bone graft substitutes intended to address the key elements of bone regeneration. Bone graft substitutes are composed of natural biologic proteins and synthetic materials. They are designed to reduce the amount of autologous bone grafts needed for spinal fusion procedures. Bone graft substitutes, depending on their design, can be used entirely in place of the patient’s own bone tissue, called an autograft, or by extending the volume of bone graft material from the patient by combining it with the bone graft substitute. Our Biologics portfolio includes fibers-based and particulate DBM, collagen ceramic matrices, demineralized cancellous allograft bone and synthetic bone void fillers. We offer our Biologics products in the form of fibers, putties, pastes, strips and DBM in a resorbable mesh for a range of surgical applications.
Spinal Implants and Enabling Technologies
Our Spinal Implants and Enabling Technology portfolio consists of an extensive line of products for spinal decompression, alignment, stabilization and image-guided surgical solutions as well as a surgical navigation system designed for broad spectrum use throughout the entire spinal column. Such products are typically used to facilitate fusion in degenerative, minimally invasive, and complex spinal deformity procedures throughout the lumbar, thoracic and cervical regions of the spine. Our products are increasingly focused on restoring adequate spinal balance and profile in the sagittal (front to back) plane, which we believe is widely recognized as an important factor to improve the quality of life in patients undergoing surgery for spinal degeneration or deformity.
The following table and discussion identifies our SeaSpine principal products by trade name and describes their primary applications:
| | |
Product | | Primary Application |
| |
Biologics Products | | |
| | |
Accell Bone Matrix | | An open structured, dispersed form of DBM, which increases the bioavailability of bone proteins at an earlier time in the healing cascade; when combined with traditional DBM, both fibers and particulate forms, provides a biphasic release of growth factors to promote healing |
| |
OsteoStrand Plus / OsteoStrand | | 100% Demineralized Bone Fibers product lines designed to facilitate and aid in fusion by maximizing osteoinductive content while providing an improved conductive matrix; OsteoStrand Plus incorporates our proprietary Accell Bone Matrix |
| |
Evo3/Evo3c DBM Putties | | Advanced DBM putties that combine traditional DBMs with Accell, with and without cancellous chips. |
| | |
OsteoTorrent/OsteoTorrent C | | Advanced DBM putties that combine Accell Bone Matrix and particulate DBM, with and without cancellous chips; packaged and sterilized in a dry state to improve product’s osteoinductive potential, shelf-life stability, and shelf-life |
| | |
OsteoBallast and Ballast DBM in Resorbable Mesh | | A resorbable mesh containing 100% DBM without a carrier, designed to simplify graft placement and help prevent graft migration while maximizing DBM content |
| | |
OsteoStrux and Mozaik | | Blend of collagen and β-TCP to create an osteoconductive material for bone regeneration; available in both putty and strip configurations |
| | |
Spinal Implants and Enabling Technologies Products |
| | |
Reef-TO, Reef-TA and Reef-TH interbody devices | | PEEK interbody devices featuring NanoMetalene surface technology for PLIF and TLIF procedures |
| | |
Vu a∙POD Prime NanoMetalene and Reef-A interbody devices | | PEEK interbody devices featuring NanoMetalene surface technology for ALIF procedures |
| | |
14
| | |
Regatta NanoMetalene Lateral System | | A comprehensive lateral lumbar interbody system that can be used to fuse the spine through a lateral approach |
| | |
Cambria NanoMetalene interbody device | | Interbody device used to fuse the cervical spine through an anterior approach |
| | |
Shoreline Anterior Cervical Standalone System, featuring the NanoMetalene with Reef Topography | | A modular plate and interbody device designed to maximize intraoperative flexibility to address a wide range of anatomy, surgical situations or bone in anterior cervical fusions |
| | |
Waveform | | 3D-printed interbody fusion devices for anterior cervical, transforaminal lumbar, lateral lumbar and articulating transforaminal lumbar interbody fusion |
| | |
Explorer TO expandable interbody device system | | An expandable interbody device system with complementary lordotic and parallel expanding implant options |
| | |
NorthStar OCT Posterior Cervical Fixation System | | Spinal fixation system with novel instrumentation and anatomically designed implants to provide a safe and effective solution designed to improve surgical flow when navigating through complex cervical procedures |
| |
Admiral Anterior Cervical Plating System ("ACP") | | A comprehensive and complete anterior cervical plating system designed to strike the optimal balance between strength, profile, and construct rigidity |
| | |
Mariner Posterior Fixation System | | Pedicle screw system for open and MIS procedures and adult deformity procedures featuring modular threaded technology and accompanying instrumentation designed to reduce the number of trays needed for surgery and that provides surgeons with multiple intra-operative options to facilitate posterior lumbar fixation |
| | |
NewPort MIS System | | MIS system with extended tabs for a small incision profile that offers two rod delivery options for both mini-open and percutaneous approaches |
| |
Mariner MIS Posterior Fixation System | | MIS system with low-profile, robust towers for rod introduction and reduction as well as ultra-tough modular extended tab heads, capable of providing powerful instrumented compression and distraction of the spine |
| |
Daytona Deformity System | | Complex spinal deformity procedure system that uses extended tab uniplanar and polyaxial screws with multiple rod options and intuitive instrumentation to create a versatile system adaptable to surgeon preference |
| | |
Daytona Small Stature System | | System designed to address standard to complex deformity cases in smaller-sized patients who need a lower profile construct due to anatomy constraints |
| | |
Mariner Outrigger Revision System | | An adjunct to the Mariner Posterior Fixation System designed to effectively revise and extend previous fusions |
| | |
FLASH Navigation with 7D Technology (Spine) | | A machine-vision navigation platform for use in open and mini-open posterior spinal procedures that uses proprietary visible light technology coupled with advanced software algorithms to deliver a fast, efficient, cost-effective, and radiation free solution for spine surgery |
| | |
FLASH Navigation with 7D Technology (Percutaneous) | | A valuable enhancement to the FLASH Navigation platform to address percutaneous spinal procedures; the camera-based technology coupled with 7D Machine Vision algorithms maintain the same fast, accurate, and efficient surgical workflow as the Spine platform, while also providing an imaging agnostic solution to percutaneous posterior spine surgery |
| | |
FLASH Navigation with 7D Technology (Cranial) | | A module on the FLASH Navigation platform that utilizes 7D Machine Vision Technology for cranial surgery; the visible light technology allows for a completely contactless workflow, acquires hundreds of thousands of virtual fiducials using the patient’s own anatomy, and results in nearly instantaneous cranial registrations to the skin or skull in almost any surgical position |
15
Enabling Technologies
Our machine vision FLASH navigation platform is used in a variety of posterior spinal procedures, including degenerative, deformity, tumor, trauma, and revision surgery. The platform can be utilized in MIS/Percutaneous, Mini-Open, or Open techniques. The technology also offers a comprehensive cranial platform for use in cranial neurosurgery.
Our innovative FLASH Navigation System with 7D Technology delivers a comprehensive navigation platform that utilizes visible light, machine-vision cameras, and intelligent software algorithms to create a 3D image within seconds for surgical navigation. The novel technology allows for a fast image reconstruction for surgical navigation with no disruption to surgeon workflow and eliminates radiation exposure during the procedure to the patient, surgeon, and operating room staff.
Our Spine Module is our leading product in the FLASH Navigation Portfolio with over 104 installations globally. In 2022, we further enhanced the Spine Module by adding preplanning features, as well as fully integrating the Mariner Posterior Fixation System, Mariner MIS Posterior Fixation System, and the Northstar OCT Posterior Cervical Fixation System into the platform with both hardware and software enhancements. We also released our commercial FLASH Percutaneous Spine Module in 2022 for the navigation of minimally invasive spinal procedures. This application, accompanied by new instrumentation, addresses an important part of the spine navigation market to round out the FLASH Navigation Platform and is a valuable enhancement for both hospitals and ambulatory surgery centers. Further enhancements and new features to the Spine Module and Percutaneous Module are in development and are expected to launch in 2023.
In addition to these new products focused on spine, the FLASH Navigation Portfolio also consists of our Cranial Module for use in cranial surgeries. The technology uses a completely contactless workflow, acquiring hundreds of thousands of virtual fiducials using the patient’s own anatomy, and results in nearly instantaneous cranial registrations to the skin or skull in almost any surgical position. New developments are also underway and expected to launch in 2023 which leverage the 7D Technology to further expand cranial applications and enter the neurocritical care market with the launch of FLASH EVD ("Extra Ventricular Drainage"), a mobile bed-side navigational system designed for fast and reliable EVD placement.
SeaSpine Future Product Applications and Development
We believe that our future success and ability to continue to drive revenue growth depends on our ability to sustain a similar cadence of launching new and next-generation products as we have demonstrated over the last few years. We continue to aggressively develop differentiated new products that we believe will allow the entrance into new markets and be even more competitive in markets in which we are underrepresented.
We expect to launch the next iteration of the FLASH Percutaneous Module and FLASH Spine Module with additional enhancements to our preplanning software as well as developing the framework for navigating interbody procedures. We also plan to launch FLASH EVD, a small mobile bed-side navigational system designed for fast and reliable EVD placement that will expand our total addressable market with this first entry into the neurocritical care market.
Product Development
Our primary research and development facilities are located in Lewisville, Texas, Carlsbad, California, Toronto, Canada, and Verona, Italy.
We have a research and development organization dedicated to advancing our portfolio of spinal implants, biologics, orthopedic implants and external fixation devices, and machine vision image guidance innovations through product development and clinical affairs programs. Our product development efforts employ an integrated team approach that involves collaboration between surgeons, our engineers, our machinists, as well as our regulatory personnel. We also work with leading hospital research institutions and certain non-profit organization, such as MTF Biologics, surgeons, and other consultants, on the long-term scientific planning and evolution of our products and therapies. Several of the products that we market have been developed through these collaborations. In addition, we periodically receive suggestions for new products and product enhancements from the scientific and medical community, some of which result in us entering into assignment or license agreements with physicians and third parties.
For our spine and orthopedics products, our product development teams, in consultation with design surgeons, formulate a design for the product and then our machinists build prototypes for testing our prototyping development and testing operation at our facilities. We use a broad scope of technologies designed to allow us to meet the complex engineering requirements of customers. As part of the development process, surgeons test the implantation of the products in our in-house cadaveric laboratories, which helps us design new products intended to meet the needs of both the surgeon and the patient. Our team refines or redesigns the prototype as necessary based on the results of the product testing, allowing us to perform rapid iterations of the design-prototype-test development cycle. Our clinical and regulatory personnel work in parallel with our product engineering personnel to facilitate
16
regulatory clearances of our products. We believe that these product development efforts allow us to provide solutions that respond to the needs of our surgeon customers and their patients.
Similar to the spine and orthopedics product development process, our software engineers, product managers and design surgeons are working towards the full integration of our spinal implants and biologics product lines with our machine vision FLASH navigation system. This includes the design of specific software modules, features and tracked instruments designed to meet the needs of a wide range of procedures including, degenerative, complex, revision, and deformity spine procedures. In addition, we are also exploring opportunities to integrate the 7D Technology into a variety of adult and pediatric orthopedic applications.
For biologics, we plan to develop line extensions for our innovative biologics technologies that will continue to improve bone forming potential while addressing specific procedural requirements both in the spine field and in general orthopedic applications. We are investigating new product formulations in the traditional DBM and Ceramic Matrix product categories. Our Biologics research and development team has experience in biomaterial sciences and bringing next generation technologies to market.
In 2022, 2021, and 2020, we incurred research and development expenses of $49.1 million, $49.6 million, and $39.1 million, respectively.
Patents, Trade Secrets, Assignments and Licenses
We rely on a combination of patents, trade secrets, assignment and license agreements, and non-disclosure agreements to protect our proprietary intellectual property. We possess numerous U.S. and foreign patents, have numerous pending patent applications, and have license rights under patents held by third parties. Our primary products are patented in the major markets in which they are sold. We do not believe that the expiration of any single patent is likely to significantly affect our intellectual property position. The medical device industry is characterized by the existence of a large number of patents and frequent litigation based on allegations of patent infringement. Patent litigation can involve complex factual and legal questions and its outcome is uncertain. Our success is dependent, in part, on us not infringing upon patents issued to others, including our competitors and potential competitors. While we make extensive efforts to ensure that our products do not infringe other parties’ patents and proprietary rights, our products and methods may be covered by patents held by our competitors. For a further discussion of these risks, please see Item 1A of this Annual Report under the heading “Risk Factors.”
We rely on confidentiality and non-disclosure agreements with employees, consultants, and other parties to protect, in part, trade secrets and other proprietary technology.
We obtain assignments or licenses of varying durations for certain of our products from third parties. We typically acquire rights under such assignments or licenses in exchange for lump-sum payments or arrangements under which we pay a percentage of sales to the licensor. However, while assignments or licenses to us generally are irrevocable, no assurance can be given that these arrangements will continue to be made available to us on terms that are acceptable to us, or at all. The terms of our license and assignment agreements vary in length from a specified number of years, to the life of product patents, or for the economic life of the product. These agreements generally provide for royalty payments and termination rights in the event of a material breach.
Compliance and Ethics Program
It is our fundamental policy to conduct business in accordance with the highest ethical and legal standards. We have a comprehensive compliance and ethics program, which is overseen by a Chief Ethics and Compliance Officer, who reports directly to our Chief Executive Officer and the Compliance Committee of the Board of Directors. The program is intended to promote lawful and ethical business practices throughout our domestic and international businesses. It is designed to prevent and detect violations of applicable federal, state, and local laws in accordance with the standards set forth in guidance issued by the U.S. Department of Justice (“U.S. DOJ”) (“Evaluation of Corporate Compliance Programs” (updated June 2020)), the Office of Inspector General (HCCA-OIG “Measuring Compliance Program Effectiveness: A Resource Guide” (March 2017)), and the U.S. Sentencing Commission (“Effective Compliance and Ethics Programs” (November 2014)). Key elements of the program include:
Organizational oversight by senior-level personnel responsible for the compliance function within the Company
Written standards and procedures, including a Corporate Code of Conduct
Methods for communicating compliance concerns, including anonymous reporting mechanisms
Investigation and remediation measures to ensure a prompt response to reported matters and timely corrective action
Compliance education and training for employees and contracted business associates
17
Auditing and monitoring controls to promote compliance with applicable laws and to assess program effectiveness
Disciplinary guidelines to enforce compliance and address violations
Due diligence reviews of high risk intermediaries and exclusion lists screening of employees and contracted business associates
Risk assessments to identify areas of compliance risk.
Government Regulation
Classification and Approval of Products by the FDA and other Regulatory Authorities
Our research, development, and clinical programs, and our manufacturing and marketing operations, are subject to extensive regulation in the U.S. and other countries. Most notably, all of our products sold in the U.S. are subject to the Federal Food, Drug, and Cosmetic Act (the “FDCA”) and the Public Health Services Act as implemented and enforced by the FDA. The regulations that cover our products and facilities vary widely from country to country. The amount of time required to obtain approvals or clearances from regulatory authorities also differs from country to country.
Unless an exemption applies, each medical device we commercially distribute in the U.S. is covered by premarket notification (“510(k)”) clearance, letter to file, approval of a premarket approval application (“PMA”), or some other approval from the FDA. The FDA classifies medical devices into one of three classes, which generally determine the type of FDA approval required. Devices deemed to pose low risk are placed in class I, devices deemed to pose moderate risk are placed in class II, and devices deemed to pose the greatest risks, requiring more regulatory controls to provide a reasonable assurance of safety and effectiveness, or devices deemed not substantially equivalent to a device that previously received 510(k) clearance (as described below), are placed in class III. Our Spinal Implants and Global Orthopedics products are, for the most part, classified as class II devices and the instruments used with these products are generally classified as class I. Our 7D FLASH navigation system is classified as class II and certain accessories thereto are classified as class I. Our Bone Growth Therapies products and the M6-C artificial cervical disc are currently classified as class III, and have been approved for commercial distribution in the U.S. through the PMA process. However, an FDA panel recommended that bone growth stimulator devices be reclassified by the FDA from class III to class II devices with special controls. For additional discussion of this development, see Item 1A of this Annual Report under the heading “Risk Factors.”
The medical devices we develop, manufacture, distribute, and market are subject to rigorous regulation by the FDA and numerous other federal, state, and foreign governmental authorities. The process of obtaining FDA clearance and other regulatory approvals to develop and market a medical device, particularly from the FDA, can be costly and time-consuming, and there can be no assurance such approvals will be granted on a timely basis, if at all. While we believe we have obtained all necessary clearances and approvals for the manufacture and sale of our products and that they are in material compliance with applicable FDA and other material regulatory requirements, there can be no assurance that we will be able to continue such compliance.
In 2017, the European Union (“E.U.”) adopted the E.U. Medical Device Regulation (“MDR”) (Council Regulations 2017/745), which imposes strict requirements for the marketing and sale of medical devices, including new quality system and post-market surveillance requirements. The regulation, as amended in March 2023, provides a transition period for all currently-approved medical devices prior to May 2021 (under the European Medical Device Directive) to meet the additional requirements, and for certain devices, this transition period was extended until December 2027 for higher risk devices and until December 2028 for medium-and-lower risk devices. After this transition period, all medical devices marketed in the E.U. will require certification according to these new requirements. This regulation has required us to incur, and we expect to continue to incur, significant costs through the transition period and beyond to maintain compliance with the additional requirements. Failure to meet the requirements of the regulation could adversely impact our business in the E.U. and other countries that utilize or rely on E.U. requirements for medical device registrations.
In the E.U., our products that contain human-derived tissue, including demineralized bone material, are not medical devices as defined in the MDR. They are also not medicinal products as defined in Directive 2001/83/EC of the European Parliament and of the Council of the E.U. Today, the regulations in the E.U. governing products that contain human-derived tissue, if applicable, vary from one E.U. member state to the next. Because of the absence of a harmonized regulatory framework and the proposed regulation for advanced therapy medicinal products in the E.U., the approval process for human-derived cell or tissue-based medical products in the E.U. may be extensive, lengthy, expensive, and unpredictable.
Certain countries, as well as the E.U., have issued regulations that govern products that contain materials derived from animal sources. Regulatory authorities are particularly concerned with materials infected with the agent that causes bovine spongiform encephalopathy ("BSE"). These regulations affect our biomaterial products for the spine, which contain material derived from bovine
18
tissue. Although we take steps designed to provide that our products are safe and free of agents that can cause disease, products that contain materials derived from animals, including our products, may become subject to additional regulation, or even be banned in certain countries, because of concern over the potential for prion transmission. Significant new regulations, a ban of our products, or a movement away from bovine-derived products because of an outbreak of BSE could have a material and adverse effect on our business or our ability to expand our business. See “Risk Factors-Risks Related to Non-Compliance with Laws and Regulations - Certain of our products contain materials derived from animal sources and may become subject to additional regulation.”
Within our Biologics product category, we market tissue for bone repair and reconstruction under the brand name Trinity ELITE, our allogeneic bone matrix comprised of cancellous bone containing viable cells and a demineralized cortical bone component. In addition, we provide demineralized cortical fiber technologies under the brand name FiberFuse, structural allografts for spinal fusion, and an amniotic membrane, which is a natural tissue barrier. These allografts are regulated under the FDA’s Human Cell, Tissues and Cellular and Tissue-Based Products ("HCT/P") regulatory paradigm and not as a medical device, biologic, or a drug. These tissues are regulated by the FDA as minimally-manipulated tissue and are covered by the FDA’s “Good Tissues Practices” regulations, which cover all stages of allograft processing. There can be no assurance our suppliers will continue to meet applicable regulatory requirements or that those requirements will not be changed in ways that could adversely affect our business. Further, there can be no assurance these products will continue to be made available to us or that applicable regulatory standards will be met or remain unchanged. Moreover, products derived from human tissue or bones are from time to time subject to recall for certain administrative or safety reasons and we may be affected by one or more such recalls.
In addition to our allograft solutions (HCT/Ps), we market and distribute additional biologics products that are synthetic in nature and are regulated by the FDA as medical devices, specifically Opus BA and the Opus MG lines of synthetic grafts. We also provide ancillary technologies regulated by the FDA as medical devices that aid in the delivery of our bone grafting options clinically. These products are sourced from third party manufacturers, which we believe maintain an adequate inventory to avoid disruptions in product supply.
We also manufacture products derived from human tissue (demineralized bone tissue). Internally produced HCT/Ps may fall within the definition of a biological product, medical device, or drug regulated under the FDCA. These biologic, device or drug HCT/Ps must comply both with the requirements exclusively applicable to HCT/Ps and with requirements applicable to biologics, devices or drugs, including premarket clearance or approval from the FDA.
Section 361 of the Public Health Service Act authorizes the FDA to issue regulations to prevent the introduction, transmission, or spread of communicable disease. HCT/Ps regulated as 361 HCT/Ps are subject to requirements relating to registering facilities and listing products with the FDA, screening and testing for tissue donor eligibility, Good Tissue Practice when processing, storing, labeling, and distributing HCT/Ps, including required labeling information, stringent record keeping, and adverse event reporting.
The American Association of Tissue Banks ("AATB") has issued operating standards for tissue banking. Accreditation is voluntary, but compliance with these standards is a requirement to become an AATB-accredited tissue establishment. In addition, some states in the U.S. have their own tissue banking regulations. We are AATB-accredited and licensed or have permits for tissue banking in California, Florida, New York, Maryland, and other states that require specific licensing or registration.
Procurement of certain human organs and tissue for transplantation is subject to the restrictions of the National Organ Transplant Act (NOTA), which prohibits the transfer of certain human organs, including skin and related tissue for valuable consideration, but permits the reasonable payment associated with the removal, transportation, implantation, processing, preservation, quality control and storage of human tissue and skin. We reimburse tissue banks for their expenses associated with the recovery, storage and transportation of donated human tissue they provide to us for processing. We include in our pricing structure amounts paid to tissue banks to reimburse them for their expenses associated with the recovery and transportation of the tissue, in addition to certain costs associated with the processing, preservation, quality control and storage of the tissue, marketing and medical education expenses, and costs associated with development of tissue processing technologies. NOTA payment allowances may be interpreted to limit the amount of costs and expenses that we may recover in our pricing for our products, thereby reducing our future revenue and profitability.
For a further description of some of the risks associated with matters described above, see Item 1A of this Annual Report under the heading “Risk Factors.”
Certain Other Product and Manufacturing Regulations
After a device is placed in the market, numerous regulatory requirements continue to apply. These regulatory requirements include: product listing and establishment registration; Quality System Regulation (“QSR”), which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation, and other quality assurance procedures during all aspects of the manufacturing process; labeling regulations and governmental prohibitions against the promotion of products for
19
uncleared, unapproved, or off-label uses or indications; clearance of product modifications that could significantly affect safety or efficacy or that would constitute a major change in intended use of one of our cleared devices; approval of product modifications that affect the safety or effectiveness of one of our PMA approved devices; Medical Device Adverse Event Reporting regulations, which require that manufacturers report to the FDA and other foreign governmental agencies if their device may have caused or contributed to a death or serious injury, or has malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction of the device or a similar device were to recur; post-approval restrictions or conditions, including post-approval study commitments; post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device; the FDA’s recall authority, whereby it can ask, or under certain conditions, order device manufacturers to recall a product from the market that is in violation of governing laws and regulations; regulations pertaining to voluntary recalls; and notices of corrections or removals.
We and certain of our suppliers also are subject to announced and unannounced inspections by the FDA and European Notified Bodies to determine our compliance with the FDA’s QSR and other international regulations. If the FDA were to find that we or certain of our suppliers have failed to comply with applicable regulations, the agency could institute a wide variety of enforcement actions, ranging from a public warning letter to more severe sanctions, such as: fines and civil penalties against us, our officers, our employees, or our suppliers; delays in clearing or approving, or refusal to clear or approve our products; withdrawal or suspension of approval of our products or those of our third-party suppliers by the FDA or other regulatory bodies; product recall or seizure; interruption of production; operating restrictions; injunctions; and criminal prosecution. In addition to FDA inspections, all of our manufacturing facilities are subject to annual Notified Body inspections.
Moreover, governmental authorities outside the U.S. have become increasingly stringent in their regulation of medical devices. Our products may become subject to more rigorous regulation by non-U.S. governmental authorities in the future. Additional regulation, whether in the U.S. or internationally, may have a material adverse effect on our business and operations. For a description of some of the risks associated with the regulatory requirements described above, see Item 1A of this Annual Report under the heading “Risk Factors.”
Accreditation Requirements
Our subsidiary, Orthofix US LLC, has been accredited by the Accreditation Commission for Health Care, Inc. (“ACHC”), for medical supply provider services with respect to durable medical equipment, prosthetics, orthotics, and supplies (“DMEPOS”). ACHC, a private, not-for-profit corporation, which is certified to ISO 9001:2000 standards, was developed by home care and community-based providers to help companies improve business operations and quality of patient care. Although accreditation is generally a voluntary activity, where healthcare organizations submit to peer review their internal policies, processes, and patient care delivery against national standards, the Centers for Medicare and Medicaid Services (“CMS”) required DMEPOS suppliers to become accredited. We believe that by attaining accreditation, Orthofix US LLC has demonstrated its commitment to maintain a higher level of competency and a willingness to strive for excellence in its products, services, and customer satisfaction.
Third-Party Payor Requirements
Our products may be reimbursed by third-party payors, such as government programs, including Medicare, Medicaid, and Tricare, or private insurance plans and healthcare networks. Third-party payors may deny reimbursement if they determine that a device provided to a patient or used in a procedure does not meet applicable payment criteria or if the policyholder’s healthcare insurance benefits are limited. Also, non-government third-party payors are increasingly challenging the medical necessity and prices paid for our products and services. The Medicare program is expected to continue to implement a new payment mechanism for certain DMEPOS items via the implementation of its competitive bidding program. Bone growth therapy devices are currently exempt from this competitive bidding process.
Laws Regulating Healthcare Fraud and Abuse; State Healthcare Laws
Our sales and marketing practices are also subject to a number of U.S. laws regulating healthcare fraud and abuse, such as the federal Anti-Kickback Statute and the federal Physician Self-Referral Law (known as the “Stark Law”), the Civil False Claims Act, and the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), as well as numerous state laws regulating healthcare and insurance. These laws are enforced by the Office of Inspector General within the U.S. Department of Health and Human Services (“HHS”), the U.S. DOJ, and other federal, state, and local agencies. Among other things, these laws and others generally (i) prohibit the provision of anything of value in exchange for the referral of patients or for the purchase, order, or recommendation of any item or service reimbursed by a federal healthcare program (including Medicare and Medicaid); (ii) require that claims for payment submitted to federal healthcare programs be truthful; (iii) prohibit the transmission of protected healthcare information to persons not authorized to receive that information; and (iv) require the maintenance of certain government licenses and permits.
20
Laws Protecting the Confidentiality of Health Information
U.S. federal and state laws protect the confidentiality of certain health information, in particular individually identifiable information such as medical records, and restrict the use and disclosure of that protected information. At the federal level, the HHS promulgates health information privacy and security rules under HIPAA. These rules protect health information by regulating its use and disclosure, including for research and other purposes. Failure of a HIPAA “covered entity” to comply with HIPAA regarding such “protected health information” could constitute a violation of federal law, subject to civil and criminal penalties. Covered entities include healthcare providers (including certain of those that sell devices or equipment) that engage in particular electronic transactions, including, as we do, the transmission of claims to health plans. Consequently, health information that we access, collect, analyze, and otherwise use and/or disclose includes protected health information that is subject to HIPAA. As noted above, many state laws also pertain to the confidentiality of health information. Such laws are not necessarily preempted by HIPAA, in particular those state laws that afford greater privacy protection to the individual than HIPAA. These state laws typically have their own penalty provisions, which could be applied in the event of an unlawful action affecting health information.
In the E.U., the General Data Protection Regulation (“GDPR”), includes, among other things, a requirement for prompt notice of data breaches to data subjects and supervisory authorities in certain circumstances and significant fines for non-compliance. Internationally, some countries have also passed laws that require individually identifiable data on their citizens to be maintained on local servers and that may restrict transfer or processing of that data.
These laws and regulations impact the ways in which we use and manage personal data, protected health information, and our information technology systems. They also impact our ability to move, store, and access data across geographic boundaries. Compliance with these requirements may require changes in business practices, complicate our operations, and add complexity and additional management and oversight needs. They also may complicate our clinical research activities, as well as product offerings that involve transmission or use of clinical data.
Physician Payments Sunshine Provision of the Affordable Care Act
The Physician Payments Sunshine Provision of the Affordable Care Act (Section 6002) (the “Sunshine Act”), requires public disclosure to the U.S. government of payments to physicians and teaching hospitals, including in-kind transfers of value, such as gifts or meals. The Sunshine Act also provides penalties for non-compliance. The Sunshine Act requires that we file an annual report on March 31st of a calendar year for the transfers of value incurred for the prior calendar year.
In 2018, the Substance Use-Disorder Prevention that Promotes Opioid Recovery and Treatment for Patients and Communities Act (the “SUPPORT Act”) was signed into law. The SUPPORT Act expands the reporting obligation under the Sunshine Act to include payments and other transfers of value made to physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists, and certified nurse midwives. These expanded reporting obligations were effective for payments reported in 2022, with payment tracking beginning in 2021. Non-compliance with the Sunshine Act or SUPPORT Act is subject to civil monetary penalties.
In addition to the Sunshine Act, as expanded by the SUPPORT Act, we seek to comply with other international and individual state transparency laws, such as the transparency laws of Massachusetts and Vermont.
Sales, Marketing and Distribution
We have a broad sales network comprised of direct sales representatives, sales agents, and distributors. This established sales network provides us with a platform to introduce new products and expand sales of existing products. Our products are distributed in approximately 68 countries worldwide.
Reporting Segments and Product Categories
Historically, Orthofix has managed the business by two reporting segments, Global Spine and Global Orthopedics, which account for 77% and 23%, respectively, of our total net sales in 2022. Comparatively, SeaSpine has historically managed its business as one operating segment, but with revenue reported in two product categories: (i) Biologics (formerly recognized as Orthobiologics) and (ii) Spinal Implants and Enabling Technologies. Following the merger with SeaSpine, which was completed on January 5, 2023, we
21
expect to reassess our reporting segments in the first quarter of 2023 based on how the operations of the newly combined company will be managed.
Sales Network
Our U.S. sales network is generally comprised of a mix of direct sales representatives and independent distributors, dependent upon each product category. An increasing number of these independent distributors sell products for more than one product category. Our Bone Growth Therapies product category is largely supported by a hybrid distribution network of direct sales representatives and independent distributors, whereas our Spinal Implants, Biologics, and Orthopedics sales organizations primarily consist of regional and territory business managers who oversee a broad network of independent distributors and sales agents.
We market our Enabling Technologies portfolio through a direct sales force in the U.S. that works together with our independent sales agents to generate either a capital sale or to place systems and components in an account in a capital efficient manner in return for a long-term revenue commitment for our spine and/or biologics products.
In the U.S., we typically consign our Biologics products and consign or loan our Spinal Implants and Orthopedics implant sets to hospitals and independent sales agents, who in turn deliver them to the hospital for a single surgical procedure or leave them with hospitals that are high volume users for use in multiple procedures. These sets typically contain the instruments, including disposables, and implants required to complete a surgery.
We focus on entering distribution relationships in territories with a high potential for growth, where our partner will carry our products exclusively, except with respect to clinical markets that our products do not address. We believe these more exclusive relationships allow us to grow faster and more cost effectively in these territories over the long term. We also plan to continue to invest in additional instrument sets and marketing and education efforts to support the expansion of our independent sales agent footprint.
Outside the U.S., we employ direct sales representatives in certain markets and also contract with independent stocking distributors, who purchase our products directly from us and independently sell them. In order to provide support to our independent sales network, we have sales and product specialists who regularly visit independent distributors to provide training and product support.
Marketing and Product Education
We market and sell our products principally to physicians, hospitals, ambulatory surgery centers, integrated health delivery systems, and other purchasing organizations.
We support our sales force and sales expansion efforts through comprehensive and specialized training workshops for physicians and sales specialists consistent with the AdvaMed Code of Ethics (“AdvaMed Code”) and the MedTech Europe Code of Ethical Business Practice (“MedTech Code”). We organize regular multilingual teaching seminars in multiple locations and also virtually. To this end, we leverage the capacity of our hands-on cadaveric training laboratories located at our Lewisville, Texas, Carlsbad, California, and Wayne, Pennsylvania facilities to increase the number of training opportunities for surgeons and sales agents. In-person trainings are also held at our facility in Verona, Italy, and in various locations in Latin America. We believe training and education will help surgeons become adept with our products and techniques, thereby improving outcomes for their patients. In recent years, thousands of surgeons from around the world have attended these in person and virtual product education seminars, which have included a variety of lectures from specialists, as well as demonstrations and hands-on workshops.
22
We also produce marketing and training materials, including materials outlining surgical procedures, for our customers, sales force, and distributors in a variety of languages using printed, video, and multimedia formats. We require all of our sales force, direct and independent, to undergo extensive product, policy, and compliance training to ensure adherence to our standards, policies, and applicable law.
Competition
The global spine, biologics, orthopedics, and image guided surgery markets are highly competitive. We face significant competition in these markets from the spine and orthopedic divisions of large multinational medical device companies, established companies focused solely or primarily on spine and orthopedics, and from smaller, emerging companies focused on product innovation. These competitors are focused on bringing new technologies to market and acquiring technologies and technology licenses that directly compete with our products or that have potential product advantages that could render our products obsolete or noncompetitive.
Our Bone Growth Therapies product category competes principally with similar products marketed by Zimmer Biomet, DJO Global, and Bioventus. Our primary competitors in the Biologics, Enabling Technologies, and Spinal Implants markets include Alphatec Spine, Baxter, B. Braun, Brainlab, Bioventus, Cerapedics, DePuy Synthes Spine (a Johnson & Johnson company), Globus Medical, Medtronic, NuVasive, Stryker, Surgalign, XTANT Medical, ZimVie and various smaller public and private companies. For Global Orthopedics devices, our principal competitors include DePuy Synthes, Zimmer Biomet, Stryker, Smith & Nephew, and OrthoPediatrics.
We believe that we enhance our competitive position by focusing on product features such as ease of use, versatility, cost, and patient acceptability, together with value-added services, such as the STIM onTrack mobile app, HEX RAY software, OrthoNext preoperative planning, and our medical education services. We attempt to avoid competing based solely on price. Overall cost and medical effectiveness, innovation, reliability, value-added service, and training are the most prevalent methods of competition in the markets for our products, and we believe we compete effectively.
Manufacturing and Sources of Supply
In general, raw materials essential to our businesses are readily available from multiple sources. For reasons of quality assurance, availability or cost effectiveness, certain components and raw materials are available only from one supplier. Our relationships with suppliers that cannot be replaced without a material expense or delay are governed by written contracts, which are generally supply agreements. These agreements set forth the process by which we order components or raw materials, as applicable, from such suppliers (which process is either on a purchase order basis or based on quarterly or annual forecasts and in some cases require us to purchase minimum amounts) and the related fees for purchasing such components or raw materials. These agreements outline the rights of each party with respect to quality assurance, inspection and compliance with applicable law and contain what we believe to be customary indemnification provisions for commercial agreements. Each of these agreements is entered into in the ordinary course of our business, is immaterial in amount and significance, and not a contract upon which our business is substantially dependent. In addition, we endeavor to maintain sufficient inventory of components and raw materials so that our production will not be significantly disrupted even if a particular component or material is not available for a period of time.
Spine and Orthopedic Products
We generally design, develop, assemble, test, and package our bone growth stimulation, spinal implant, and orthopedic products, and subcontract the manufacturing of a substantial portion of the component parts and instruments. Although certain of our key raw materials are obtained from a single source, we believe alternate sources for these materials are available. Further, we believe an adequate inventory supply is maintained to avoid product flow interruptions. Historically, we have not experienced difficulty in obtaining the materials necessary to meet our production schedules.
Our products are currently manufactured and assembled in the U.S., Canada, and Italy. We believe our plants comply in all material respects with the requirements of the FDA and all relevant regulatory authorities outside the U.S. For a description of the laws to which we are subject, see Item 1, “Business”, under the subheadings “Corporate Compliance and Ethics Program” and “Government Regulation.” We actively monitor each of our subcontractors in order to maintain manufacturing and quality standards and product specification conformity.
Biologics
Most of our Biologics products contain material derived from human or bovine tissue. We only source our raw materials from tissue banks registered with the FDA and accredited by the AATB. The donors are screened, tested and processed by the tissue banks in accordance with FDA and AATB requirements. Additionally, each donor must pass FDA-specified bacterial and viral testing before
23
raw material is distributed to us for further processing. We receive with each donor lot a certification of the safety of the raw material from the tissue bank’s medical director. As an added safety assurance, each lot of bone is released into the manufacturing process only after our quality assurance microbiologists screen the incoming bone and serology test records. During our manufacturing process, the bone particles are subjected to our proprietary process and terminally sterilized. This process is designed to support the safety and effectiveness of our DBM products.
The collagen used in our collagen ceramic matrix products is derived only from the deep flexor tendon of cattle less than 24 months old from New Zealand. The World Health Organization classifies different types of cattle tissue for relative risk of BSE transmission. Deep flexor tendon is in the lowest-risk category for BSE transmission (the same category as milk, for example) and is therefore considered to have a negligible risk of containing the agent that causes BSE (an improperly folded protein known as a prion).
We also partner with MTF Biologics to provide our customers allograft solutions (HCT/Ps) for various spine, orthopedic and other bone repair needs. MTF Biologics provides donor screening, processing, and quality standards that are expected by our customers. Our partnership with MTF allows us to exclusively market the Virtuos Lyograph, Trinity ELITE, FiberFuse and FiberFuse Strip, and certain other tissue forms and we have a non-exclusive marketing rights for our Opus BA and Opus MG Set synthetic, biologic offerings.
Human Capital Resources
Our key human capital objectives in managing our business include attracting, developing, and retaining top talent while integrating diversity, equity, and inclusion principles and practices into our core values.
Employees
At December 31, 2022, we had 1,092 employees worldwide. Of these, 786 were employed in the U.S. and 306 were employed at other non-U.S. locations. Our relations with our Italian employees, who numbered 227 at December 31, 2022, are governed by the provisions of a National Collective Labor Agreement setting forth mandatory minimum standards for labor relations in the metal mechanic workers industry. We are not a party to any other collective bargaining agreement.
Subsequent to the merger with SeaSpine, which was completed on January 5, 2023, we have approximately 1,734 employees worldwide, with 1,371 employed in the U.S. and 363 employed at other non-U.S. locations.
Compensation and Benefits
Because attracting, developing, and retaining high-level talent is a key component of our human capital objectives, we seek to provide competitive compensation and benefits packages, and to prioritize the health and wellness our employees. In addition to the comprehensive and competitive health plans that we offer, our employees receive access to the following benefits: a 401(k) retirement plan with a Company match, an employee stock purchase plan, virtual physician consults, an employee health advocate, a Company-provided basic life insurance and disability benefits corporate wellness program, an onsite fitness center for certain locations, paid parental leave, an employee assistance program, a flexible spending account, health savings accounts, and local employee discounts programs. Through our Innovator and Above and Beyond Award programs we recognize and reward our employees that exemplify our mission of providing transformative solutions that improve patients' lives.
Talent Development
We believe that success comes from investing in our people and ensuring our workforce is aligned with our mission and values. To achieve this goal, we devote time and resources to assist our employees in being familiar with our business, industry, and product offerings. We have developed a robust onboarding program for our newly hired associates that provides a comprehensive overview of our product portfolio and company history. We put an emphasis on training our employees and sales representatives to understand our business, including the underlying medical conditions that our products treat. In addition, we strive to support our teams in the areas of development, mentoring, engagement, and health and wellness, enabling them to do their best work as they grow their careers. In 2022, we successfully completed our second annual summer internship program with 80% of participating interns meeting a diversity criteria. Additionally, in 2022 we matched interns hired from our 2021 program to employee mentors, continued our 2021 Leadership Excellence and Acceleration Program (“LEAP”) inaugural cohort, and prepared for a second cohort to launch in 2023, which will include a minimum of 25% minority participation.
24
Diversity and Inclusion
We are committed to fostering, cultivating, and preserving a culture that promotes diversity, equity, and inclusion. We seek to demonstrate our commitment to providing equal and equitable opportunities to all employees through programs such as our Moving 4ward initiative, a program created to embrace the value of diversity and reflect the communities where we live and work. Additionally, we proudly support the Orthofix Women’s Network, a program that provides opportunities for women to learn from each other and grow within our company and our industry. Throughout the year, we promote a variety of diverse voices to our employees by recognizing events such as Black History Month, Martin Luther King Jr. Day, Women’s History Month, Asian Pacific American Heritage Month, LGBTQ Pride Month, Juneteenth, and Hispanic Heritage Month. We seek to embrace and encourage our employees’ differences and know that diversity, equity and inclusion help build a truly global, transformative business and will continue to be a source of our strength. Building on this belief, we launched companywide, and incorporated into our new hire orientation, a training titled, “Hiring, Leading and Fostering Diverse and Inclusive Teams”. We intend that by end of 2023, all hiring managers, leaders, and interviewers will have completed this training.
Health and Safety
Promoting and protecting the safety of our workforce is a top priority. Health and safety matters are responsibilities that we share throughout our organization. We evolved in these matters during the last few years to meet the needs of our workforce during the COVID-19 pandemic. Employees’ safety risks vary depending on the roles they perform, and we seek to tailor our safety efforts accordingly. We periodically measure the sentiment of our employees through an employee engagement survey and share the results and action items identified from the survey with our employees.
Community
We support a variety of charitable organizations through donations, fundraising efforts, educational partnerships with colleges and universities, and local community development. Over the years, we have raised funds and awareness for veteran support groups, food and homebuilding organizations, and health-related institutions. In 2022, we added a corporate objective to our annual incentive program to encourage community volunteerism. Under this program, our employees contributed 1,988 hours to community outreach programs, which exceeded our communicated goal. We proudly supported Donate Life, relief efforts for Ukraine, Texas Scottish Rite Hospital for Children, blood drives, food pantries and other charitable initiatives in the communities we live and work in around the world.
25
Item 1A. Risk Factors
In addition to the other information contained in this Annual Report and the exhibits hereto, you should carefully consider the risks described below. These risks are not the only ones that we may face. Additional risks not presently known to us or that we currently consider immaterial may also impair our business operations. This Annual Report also contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of certain factors, including the risks faced by us described below or elsewhere in this Annual Report. Investing in our common stock involves a high degree of risk and if any of these risks or uncertainties occur, the trading price of our common stock could decline and you could lose part or all of your investment. The disclosures in this Item 1A of this Annual Report under the heading “Risk Factors” relate to the combined company subsequent to the merger unless otherwise noted.
Summary of Risk Factors
The section provides a summary of many of the risks we are exposed to in the normal course of our business activities. The summary does not contain all of the information that may be important to you, and you should read the summary together with the more detailed discussion of risks set forth following this section as well as elsewhere in this report.
•The merger of Orthofix and SeaSpine may trigger change in control or other provisions in certain distributor, customer and other agreements, any of which may have an adverse impact on the combined company’s business and results of operations .
•Uncertainties associated with the merger may cause a loss of management personnel and other key employees.
•Stockholder litigation related to the merger could negatively affect our business and operations.
•Integration of the Orthofix and SeaSpine businesses is expected to be expensive and time-intensive and we may not be able to successfully integrate the businesses and/or realize anticipated synergies and benefits in a timely manner, if at all.
•We are subject to a wide range of requirements, regulations, and laws due to our international operations and related to the medical device industry in which we operate, the violation of any of which could subject us to adverse consequences.
•Ongoing healthcare reform initiatives and changes in third-party reimbursement policies and in the healthcare industry aimed at cost containment may adversely impact our business.
•We and certain of our suppliers are subject to extensive government regulation that increases our costs and could limit our ability to market or sell our products.
•Oversight of the medical device industry might affect the way we sell medical devices and compete in the marketplace.
•An FDA panel recommended that bone growth stimulator devices be reclassified by the FDA from Class III to Class II devices, which could increase future competition for us in this product category and negatively affect our future sales of such products.
•We are subject to requirements relating to hazardous materials which may impose significant compliance or other costs on us.
•The COVID-19 pandemic, and the related effects thereof, has materially adversely affected, and could continue to materially adversely affect, our operations, supply chain, manufacturing, product demand, product distribution, customers and other business activities.
•The ongoing conflict between Russia and Ukraine, and the global response to it, could adversely impact our global operations.
•Our business may be adversely affected if consolidation in the healthcare industry leads to demand for price concessions or if a group purchasing organization (“GPO”) or similar entity excludes us from being a supplier.
•The industry in which we operate is highly competitive. New product developments and improvements by our competitors could make our products or technologies non-competitive or obsolete. Similarly, unless clinical studies demonstrate the safety and efficacy of our products, alone and relative to competitive products, our sales may be adversely affected.
•Our ability to market products successfully depends, in part, upon the acceptance of the products not only by consumers, but also by independent third parties, including physicians, hospitals, and third party payors.
•Clinical development is a lengthy and expensive process with an inherently uncertain outcome. Failure to successfully complete clinical trials and obtain regulatory approval for our product candidates on our anticipated timelines at
26
reasonable costs to us, or at all, could have a material adverse effect on our business, operating results and financial condition.
•If the third parties on which we rely to conduct our clinical studies do not perform as contractually required or expected, we may not obtain required approvals for or commercialize our products.
•Certain of our products are derived from human tissue or contain materials derived from animal sources and are or could be subject to additional regulations.
•Unfavorable negative publicity concerning both alleged improper methods of tissue recovery from donors and disease transmission from donated tissue could limit widespread acceptance of some of our products.
•We may not be able to successfully introduce new products to the market and, if we do, market acceptance or the market size for our products may not be as we expect.
•There is no guarantee that regulatory authorities, U.S. or foreign, will grant clearance or premarket approval of our future products.
•Our success depends on our ability to successfully educate and train surgeons and their staff on the benefits, safety, cost-effectiveness, and proper use of our products.
•Security breaches, cyber-attacks, loss of data, and other disruptions to our information technology systems could compromise sensitive information and/or adversely affect our business.
•Our business could be harmed if any of our manufacturing, development or research facilities are damaged and/or our manufacturing processes are interrupted.
•We depend on a limited number of third-party manufacturers and suppliers for manufacturing and processing activities, components, and raw materials. Failure of these third parties to perform as expected could result in substantial delays, increased costs or failures of our product development programs, or delayed or unsuccessful commercialization of our products.
•We may not maintain or grow our revenue if we are unable to maintain and expand our network of independent sales representatives and distributors.
•Our success depends on the services of key members of our senior management and other key employees.
•Our business is subject to economic, political, regulatory, and other risks associated with international sales and operations.
•Our failure to adequately protect or enforce our intellectual property rights could harm our position in the marketplace or prevent or impede the commercial protection of our products.
•We may be subject to third parties claims for infringement or misappropriation of their intellectual property.
•There have been substantial intellectual property disputes in our industry, which are inherently costly, divert significant time and other resources, and have unpredictable outcomes.
•We may have significant product or other liability exposure, some of which may not be covered by insurance, and if covered by insurance, such coverage may not cover all claims, which could require us to pay substantial sums.
•Our efforts to identify, pursue, and implement new business opportunities (including acquisitions) may be unsuccessful.
•We have invested in and provided loans to privately-held companies and if they are unsuccessful, we may lose all of our investment and our loans may not be repaid.
•Our sales volumes and our operating results may fluctuate.
•Our goodwill, intangible assets and fixed assets are subject to potential impairment which could adversely affect our future financial results.
•We maintain a $300.0 million secured revolving credit facility secured by a pledge of substantially all of our property. Our failure to comply with the facility’s covenants could result in an event of default, which could adversely affect our future.
•We must maintain high levels of inventory, which could consume a significant amount of our resources and reduce our cash flows.
•Our future capital needs are uncertain and we may need to raise additional funds in the future, and such funds may not be available on acceptable terms or at all.
•Our business could be negatively impacted by corporate citizenship and environmental, social, and governance ("ESG") matters and/or our reporting of such matters.
27
Risks Related to our Recently Completed Merger with SeaSpine
The merger may trigger change in control or other provisions in certain distributor, customer and other agreements to which Orthofix or SeaSpine is a party, which may have an adverse impact on the combined company’s business and results of operations following completion of the merger.
The merger may trigger change in control and other provisions in certain agreements to which Orthofix or SeaSpine is a party. If Orthofix or SeaSpine is unable to negotiate waivers of those provisions, counterparties may exercise their rights and remedies under the agreements, including terminating the agreements or seeking monetary damages or equitable remedies. Even if Orthofix and SeaSpine are able to negotiate consents or waivers, the counterparties may require a fee for such waivers or seek to renegotiate the agreements on terms less favorable to Orthofix or SeaSpine. Any of the foregoing or similar developments may have an adverse impact on the combined company’s business and results of operations following the completion of the merger.
Uncertainties associated with the merger may cause a loss of management personnel and other key employees, which could adversely affect the future business and operations of the combined company following completion of the merger.
We are dependent on the experience and industry knowledge of our officers and other key employees to execute our business plans. The combined company’s success after the completion of the merger will depend in part upon the ability of the combined company to retain certain key management personnel and employees of Orthofix and SeaSpine. As a result of the merger, current and prospective employees may experience uncertainty about their roles following the completion of the transactions, which may have an adverse effect on our ability to attract or retain key management and other key personnel. In addition, no assurance can be given that the combined company will be able to attract or retain key management personnel and other key employees to the same extent that Orthofix and SeaSpine have previously been able to attract or retain their own employees.
Stockholder litigation could negatively affect our business and operations.
On each of November 17, 2022, November 21, 2022, and December 13, 2022, purported then-stockholders of SeaSpine filed a complaint against SeaSpine and the then-members of SeaSpine’s board of directors in the United States District Court for the Southern District of New York and in the United States District Court for the District of Delaware. In addition, on December 13, 2022, a purported then-stockholder of Orthofix filed a complaint against Orthofix and the then-members of Orthofix’s board of directors in the United States District Court for the Southern District of New York. The complaints assert claims under Section 14(a) of the Exchange Act and Rule 14a-9 promulgated thereunder and Section 20(a) of the Exchange Act for allegedly causing a materially incomplete and misleading registration statement on Form S-4 filed with the SEC on November 8, 2022, or for allegedly causing a materially incomplete and misleading Schedule 14A definitive proxy statement filed with the SEC on November 23, 2022. Among other remedies, the plaintiffs sought to enjoin the merger. All four of these actions have now been voluntarily dismissed by the plaintiffs. On November 19, 2022, counsel to two different purported then-stockholders of SeaSpine sent demand letters making similar assertions. On November 23, 2022, counsel to another purported then-stockholder of SeaSpine sent a draft federal court complaint containing similar allegations, making similar claims under Section 14(a) of the Exchange Act and Rule 14a-9 promulgated thereunder and Section 20(a) of the Exchange Act, and also seeking to enjoin the merger. In addition, on November 15, 2022 and December 20, 2022, counsel to two different purported then-stockholders of Orthofix sent demand letters to Orthofix’s counsel attaching draft federal court complaints against Orthofix and the then-members of the Orthofix board making similar claims under Section 14(a) of the Exchange Act and Rule 14a-9 promulgated thereunder and Section 20(a) of the Exchange Act, and also sought to enjoin the merger. On December 14, 2022, and December 22, 2022, counsel to two additional purported then-stockholders of Orthofix sent demand letters to Orthofix’s counsel containing similar allegations. Although the ultimate outcome of these lawsuits cannot be predicted with certainty, Orthofix and SeaSpine believe the claims are without merit and intend to defend against these actions vigorously.
Securities class action lawsuits and derivative lawsuits are often brought against companies that have entered into merger agreements. Additional lawsuits against Orthofix, SeaSpine, Merger Sub and/or the directors and officers of Orthofix and/or SeaSpine in connection with the merger may be filed in the future. Neither Orthofix nor SeaSpine can give assurance as to the outcome of any lawsuit that has been or may be filed, including the amount of costs associated with defending claims or any other liabilities that may be incurred in connection with such litigation. Whether or not any plaintiff’s claim is successful, this type of litigation may result in significant costs and divert management’s attention and resources, which could adversely affect the operation of Orthofix’s and SeaSpine’s business.
The combined company may be unable to successfully integrate the Orthofix and SeaSpine businesses and realize the anticipated benefits of the merger.
28
The success of the merger will depend, in part, on the combined company’s ability to successfully combine and integrate the Orthofix and SeaSpine businesses, and realize the anticipated benefits, including synergies, cost savings, innovation and technological opportunities and operational efficiencies from the merger in a manner that does not materially disrupt existing customer, supplier, and employee relations and does not result in decreased revenues due to losses of, or decreases in orders by, customers. If the combined company is unable to achieve these objectives within the anticipated time frame, or at all, the anticipated benefits may not be realized fully or at all, or may take longer to realize than expected, and the value of the combined company common stock may decline. Integration may result in additional and unforeseen expenses, and the combined company may fail to realize some or all of the anticipated benefits of the merger on a timely basis or at all.
While we have successfully completed a number of integration activities since the closing of merger, the remainder of our integration activities may not be completed smoothly or successfully. The integration of the two companies may result in material challenges, including, without limitation:
•managing a larger, more complex combined medical device business;
•maintaining employee morale and retaining key management and other employees;
•retaining existing business and operational relationships, including customers, suppliers and employees and other counterparties, as may be impacted by contracts containing consent and/or other provisions that may be triggered by the merger, and attracting new business and operational relationships;
•unanticipated issues in integrating the numerous systems involved in operating our businesses, including information technology, communications, purchasing, accounting and finance, including integrating different accounting policies, sales, billing, payroll, employee benefits, regulatory compliance and other systems;
•successfully addressing inconsistencies in standards, controls, procedures or policies that could affect our ability to maintain relationships with customers and employees or to achieve the anticipated benefits of the merger;
•consolidating corporate and administrative infrastructures and eliminating duplicative operations;
•coordinating geographically separate organizations, systems, and facilities and addressing possible differences in business backgrounds, corporate cultures, and management philosophies; and
•unforeseen expenses or delays associated with the merger.
Many of these factors will be outside of our control, and any one of them could result in delays, increased costs, decreases in the amount of expected revenues and other adverse impacts, which could materially affect the combined company’s financial position, results of operations and cash flows. In addition, the integration of certain operations requires the dedication of significant management resources, which may temporarily distract management’s attention from our day-to-day business. Employee uncertainty and lack of focus during the integration process may also disrupt our business.
In addition, SeaSpine completed its merger with 7D Surgical, Inc. in May 2021, and the integration of the SeaSpine business and 7D Surgical remains in process and remains subject to certain risks, including that (a) the benefits expected to be received from the acquisition may not be realized in their entirety, (b) there could be unanticipated adverse impacts on our or 7D Surgical’s business, and/or we may otherwise not realize the expected return on our investment, (c) we may be subject to claims or liabilities related to 7D Surgical’s business arising after the merger was completed and SeaSpine may have failed to identify or assess the magnitude of certain liabilities, shortcomings or other circumstances prior to acquiring 7D Surgical; and (d) 7D Surgical was not required to maintain an internal control infrastructure that would meet the standards of a U.S. public company, and we may incur substantial costs to implement such controls and procedures and we could encounter unexpected delays and challenges in this implementation. The ongoing integration of 7D Surgical may increase the complexity of, and challenges associated with, the integration of the Orthofix and SeaSpine businesses, which may make it more difficult for Orthofix and SeaSpine to achieve the anticipated benefits of the merger fully or at all, or within the anticipated time frame.
The future results of the combined company may be adversely impacted if the combined company does not effectively manage its complex operations following the completion of the merger.
Following the completion of the merger, the size of the combined company’s business will be significantly larger than the current size of either SeaSpine’s business or Orthofix’s business. The combined company’s ability to successfully manage this expanded business will depend, in part, upon management’s ability to design and implement strategic initiatives that address not only the integration of the Orthofix and SeaSpine businesses, but also the increased scale and scope of the combined business with its
29
associated increased costs and complexity. There can be no assurances that the combined company will be successful in integrating the businesses or that it will realize the expected operating efficiencies, cost savings and other benefits currently anticipated from the merger.
We expect to incur substantial expenses related to the completion of the merger and the integration of the Orthofix and SeaSpine businesses.
We will incur substantial expenses in connection with the completion of the merger to integrate a large number of processes, policies, procedures, operations, technologies and systems of Orthofix and SeaSpine in connection with the merger. The substantial majority of these costs will be non-recurring expenses related to the transactions and facilities and systems consolidation costs. The combined company may incur additional costs or suffer loss of business under third-party contracts that are terminated or that contain change in control or other provisions that may be triggered by the completion of the transactions, and/or losses of, or decreases in orders by, customers, and may also incur costs to retain certain key management personnel and employees. Orthofix and SeaSpine will also incur transaction fees and costs related to formulating integration plans for the combined business, and the execution of these plans may lead to additional unanticipated costs and time delays. These incremental transaction-related costs may exceed the savings the combined company expects to achieve from the elimination of duplicative costs and the realization of other efficiencies related to the integration of the businesses, particularly in the near term and in the event there are material unanticipated costs. Factors beyond the parties’ control could affect the total amount or timing of these expenses, many of which, by their nature, are difficult to estimate accurately.
The market price of the combined company common stock after the merger is completed may be affected by factors different from those affecting the price of Orthofix common stock or SeaSpine common stock before the merger is completed.
Upon completion of the merger, previous holders of Orthofix common stock and previous holders of SeaSpine common stock are now holders of common stock of the combined company. As the businesses of Orthofix and SeaSpine are different, the results of operations, as well as the price of the combined company common stock, may, in the future, be affected by factors different from those factors affecting each of Orthofix and SeaSpine as an independent stand-alone company. The combined company will face additional risks and uncertainties to which each of Orthofix and SeaSpine may not have previously been exposed. As a result, the market price of the combined company’s shares may fluctuate significantly following completion of the merger.
The market price of the combined company common stock may decline as a result of the merger, including as a result of some Orthofix and/or SeaSpine stockholders adjusting their portfolios.
The market price of the combined company common stock may decline as a result of the merger if, among other things, the operational cost savings estimates in connection with the integration of the Orthofix and SeaSpine businesses are not realized, there are unanticipated negative impacts on Orthofix’s financial position, or if the transaction costs related to the merger are greater than expected. The market price also may decline if the combined company does not achieve the perceived benefits of the merger as rapidly or to the extent anticipated by financial or industry analysts or if the effect of the transactions on the combined company’s financial position, results of operations or cash flows is not consistent with the expectations of financial or industry analysts.
In addition, sales of combined company common stock after the completion of the merger may cause the market price of such common stock to decrease. Based on the number of shares of SeaSpine common stock outstanding immediately prior to the close of the merger, Orthofix issued an aggregate of approximately 16.0 million shares of Orthofix common stock to holders of SeaSpine common stock in the merger. Historical SeaSpine stockholders may decide not to hold the shares of combined company common stock they will receive in the merger. In addition, certain Orthofix stockholders, such as funds with limitations on their permitted holdings of stock in individual issuers, may be required to sell their shares of common stock following completion of the merger. Such sales of combined company common stock could have the effect of depressing the market price for the combined company common stock.
Any of these events may (i) make it more difficult for the combined company to sell equity or equity-related securities, (ii) dilute your ownership interest in the combined company, and/or (iii) have an adverse impact on the price of the combined company common stock.
30
Risks Related to our Legal and Regulatory Environment
If we fail to maintain an effective system of internal controls or discover material weaknesses in our internal control over financial reporting, we may not be able to report our financial results accurately or detect fraud, which could harm our business and the trading price of our common stock.
Effective internal controls are necessary for us to produce reliable financial reports and are important in our effort to prevent financial fraud. We are required to periodically evaluate the effectiveness of the design and operation of our internal controls. As has occurred in several years prior, these evaluations may result in the conclusion that enhancements, modifications, or changes to our internal controls are necessary or desirable. While management evaluates the effectiveness of our internal controls on a regular basis, these controls may not always be effective. There are inherent limitations on the effectiveness of internal controls, including collusion, management override, and failure of human judgment. Because of this, control procedures are designed to reduce rather than eliminate business risks. Also, previously effective internal controls may become inadequate over time because of changes in our business or operating structure, and we may fail to take measures to evaluate the adequacy of and update these controls, as necessary. If we fail to maintain an effective system of internal controls or if management or our independent registered public accounting firm were to discover material weaknesses in our internal controls, we may be unable to produce reliable financial reports or prevent fraud, which could harm our financial condition and operating results, and could result in a loss of investor confidence and a decline in our stock price.
We are subject to the Foreign Corrupt Practices Act (the “FCPA”) and other similar anti-bribery laws and any violations of such laws could subject us to adverse consequences.
The FCPA and similar anti-bribery laws in non-U.S. jurisdictions generally prohibit companies and their intermediaries from making improper payments to foreign government officials for the purpose of obtaining or retaining business. The FCPA also imposes accounting standards and requirements on U.S. publicly traded entities and their foreign affiliates, which are intended to prevent the diversion of corporate funds to the payment of bribes and other improper payments. Because of the predominance of government-sponsored healthcare systems around the world, many of our customer relationships outside of the U.S. are with governmental entities and are therefore subject to such anti-bribery laws.
In recent years, both the U.S. and non-U.S. regulators have increased regulation, enforcement, inspections, and governmental investigations of the medical device industry, including increased U.S. government oversight and enforcement of the FCPA. Despite implementation of a comprehensive global healthcare compliance program, we may be subject to more regulation, enforcement, inspections, and investigations by governmental authorities in the future.
Any failure to comply with applicable legal and regulatory obligations in the U.S. or abroad could adversely affect us in a variety of ways that include, but are not limited to, significant criminal, civil, and administrative penalties, including imprisonment of individuals, fines and penalties, denial of export privileges, suspension or withdrawal of CE Certificates of Conformity, seizure of shipments, restrictions on certain business activities, disgorgement and other remedial measures, disruptions of our operations, and significant management distraction. Also, the failure to comply with applicable legal and regulatory obligations could result in the disruption of our distribution and sales activities. Any reduction in international sales, or our failure to further develop our international markets, could have a material adverse effect on our business, results of operations, and financial condition.
We are subject to federal and state healthcare fraud, abuse, and anti-self-referral laws, and could face substantial penalties if we are determined not to have fully complied with such laws.
Healthcare fraud and abuse regulations by federal and state governments impact our business. Healthcare fraud and abuse laws potentially applicable to our operations include:
The federal Anti-Kickback Statute, which prohibits knowingly and willfully soliciting, receiving, offering, or paying remuneration, directly or indirectly, in exchange for or to induce the purchase or recommendation of an item or service reimbursable under a federal healthcare program (such as the Medicare or Medicaid programs);
The federal Stark law, which prohibits physician self-referral, specifically a referral by a physician of a Medicare or Medicaid patient to an entity providing designated health services if the physician or an immediate family member has a financial relationship with that entity;
Federal false claims laws, which prohibit, among other things, knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other federal government payors that are false or fraudulent; and
31
State and non-U.S. laws analogous to each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by non-governmental or non-U.S. governmental third-party payors, including commercial insurers.
Federal and state government agencies, as well as private whistleblowers, have significantly increased investigations and enforcement activity under these laws. Violations of these laws are punishable by civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, or the exclusion from participation in federal, non-U.S., or state healthcare programs. Although we exercise care in structuring our sales and marketing practices, customer discount arrangements, and interactions with healthcare professionals to comply with these laws and regulations, we cannot provide assurance that government officials will not assert that our practices are not in compliance or that government regulators or courts will interpret those laws or regulations in a manner consistent with our interpretation. Even if an investigation is unsuccessful or is not fully pursued, we may spend considerable time and resources defending ourselves and the adverse publicity surrounding any assertion that we may have engaged in violative conduct could have a material and adverse effect on our reputation with existing and potential customers and on our business, financial condition, and results of operations.
Reimbursement policies of third parties, cost containment measures, and healthcare reform could adversely affect the demand for our products and limit our ability to sell our products.
Maintaining and growing sales of our products depends on the availability of adequate coverage and reimbursement from third-party payors, both within and outside the U.S. Our products are sold either directly by us or by independent sales representatives to customers or to our independent distributors and purchased by hospitals, healthcare providers, and patients. These products may be reimbursed by third-party payors, such as government programs, including Medicare, Medicaid, and Tricare, or private insurance plans, managed care organizations, and healthcare networks. Major third-party payors for medical services in the U.S. and internationally continue to work to contain health care costs, are increasingly challenging the policies and the prices charged for medical products and services, and have or may implement initiatives to limit the growth of healthcare costs, including price regulation, competitive pricing, coverage and payment policies, comparative effectiveness of therapies, technology assessments, and managed-care arrangements. Any medical policy developments that eliminate, reduce, or materially modify coverage of our reimbursement rates for our products could have an impact on our ability to sell our products. In addition, third-party payors continually review and revise their coverage and reimbursement policies for procedures involving the use of our products and can, without notice, eliminate or reduce coverage or reimbursement if they determine that a device or product provided to a patient or used in a procedure does not meet applicable payment criteria or if the policyholder’s healthcare insurance benefits are limited.
For example, in the past, a major national third-party insurer in the U.S. reduced coverage (from all or most cases to limited indications) for biomechanical devices (e.g., spine cages) used in cervical fusion procedures, stating that the devices had not been shown to be more effective than bone graft. In addition, certain insurers have limited coverage for vertebral fusions in the lumbar spine and other insurers may adopt similar coverage decisions in the future. Limits put on reimbursement could make it more difficult to buy our products and substantially reduce, or possibly eliminate, patient access to our products. In addition, should governmental authorities continue to enact legislation or adopt regulations that affect third-party coverage and reimbursement, access to our products and coverage by private or public insurers may be reduced with a consequential material adverse effect on our sales and profitability.
CMS, in its ongoing implementation of the Medicare program, periodically reviews medical study literature to determine how the literature addresses certain procedures and therapies in the Medicare population. The impact that this information could have on Medicare coverage policy for our products is currently unknown, but we cannot provide assurances that the resulting actions will not restrict Medicare coverage for our products. There can be no assurance that we or our distributors will not experience significant reimbursement problems in the future related to these or other proceedings.
As required by law, CMS has continued efforts to implement a competitive bidding program for selected DMEPOS items paid for by the Medicare program. In this program, Medicare rates are based on bid amounts for certain products in designated geographic areas, rather than the Medicare fee schedule amount. Bone growth stimulation products are currently exempt from this competitive bidding process. We cannot predict which products from any of our businesses may ultimately be affected or whether or when the competitive bidding process may be extended to our businesses. There can be no assurance that the implementation of the competitive bidding program will not have an adverse impact on the sales of some of our products.
With respect to international sales, market acceptance may depend, in part, upon the availability of coverage and reimbursement within prevailing healthcare payment systems. Reimbursement and healthcare payment systems in international markets vary significantly by country. As in the U.S., our products may not obtain coverage and reimbursement approvals in a timely manner, if at all, in a particular international market. In addition, even if we obtain country-specific coverage and reimbursement approvals, we could incur considerable expense to do so. Our failure to obtain such coverage and approvals would negatively affect market acceptance of our products in the international markets in which such failure occurs and the expenses incurred in connection with obtaining such coverage and approvals could outweigh the benefits of obtaining them.
32
Globally, our products are sold in many countries, such as the U.K., Germany, France, and Italy, which have publicly funded healthcare systems. The ability of hospitals supported by such systems to purchase our products is dependent, in part, upon public budgetary constraints. Any increase in such constraints may have a material adverse effect on our sales and collection of accounts receivable from such sales.
If the trend by governmental agencies and other third-party payors to reduce coverage of and/or reimbursement for procedures using our products continues, our business, results of operations, and financial condition could be materially and adversely affected. Further, we cannot be certain that, under current and future payment systems, the cost of our products will be adequately incorporated into the overall cost of the procedure and, accordingly, we cannot be certain that the procedures performed with our products will be reimbursed at a cost-effective level, or at all.
We and certain of our suppliers may be subject to extensive government regulation that increases our costs and could limit our ability to market or sell our products.
The medical devices we manufacture and market are subject to rigorous regulation by the FDA and numerous other federal, state, and foreign governmental authorities. These authorities regulate the development, approval, classification, testing, manufacturing, labeling, marketing, and sale of medical devices. Likewise, our use and disclosure of certain categories of health information may be subject to federal and state laws, implemented and enforced by governmental authorities that protect health information privacy and security. For a description of these regulations, see Item 1, “Business,” under the subheading “Government Regulation.”
The approval or clearance by governmental authorities, including the FDA in the U.S., is generally required before any medical devices may be marketed in the U.S. or other countries. We cannot predict whether, in the future, the U.S. or foreign governments may impose regulations that have a material adverse effect on our business, financial condition, results of operations, or cash flows.
The process of obtaining FDA clearance and approvals to develop and market a medical device can be costly, time-consuming, and subject to the risk that such clearances or approvals will not be granted on a timely basis, if at all. The regulatory process may delay or prohibit the marketing of new products and impose substantial additional costs if the FDA lengthens review times for new devices. Further, the FDA has the ability to change the regulatory classification of a cleared or approved device from a higher to a lower regulatory classification, or to reclassify an HCT/P, either of which could materially adversely impact our ability to market or sell our devices.
In addition, we must engage in extensive recordkeeping and reporting. For example, the Federal Medical Device Reporting regulation requires us to provide information to the FDA whenever there is evidence that reasonably suggests that a device may have caused or contributed to a death or serious injury or that a malfunction occurred that would be likely to cause or contribute to a death or serious injury upon recurrence.
We and certain of our suppliers also are subject to announced and unannounced inspections by the FDA to determine our compliance with FDA’s QSR and other regulations. Allegations may be made against us or against our suppliers, including donor recovery groups or tissue banks, claiming that the acquisition or processing of biomaterials products does not comply with applicable FDA regulations or other relevant statutes and regulations. Allegations like these could cause regulators or other authorities to investigate or take other action against us or our suppliers, or could cause negative publicity for us or our industry generally. If the FDA were to investigate us, because of an allegation or otherwise, and if the FDA were to conclude that we are not in compliance with applicable laws or regulations, or that any of our medical devices are ineffective or pose an unreasonable health risk, the agency could institute a wide variety of enforcement actions, ranging from a public warning letter to more severe sanctions such as fines and civil penalties against us, our officers, our employees, or our suppliers; unanticipated expenditures to address or defend such actions; delays in clearing or approving, or refusal to clear or approve, our products; withdrawal or suspension of approval of our products or those of our third-party suppliers by the FDA or other regulatory bodies; product recall or seizure; interruption of production; operating restrictions; injunctions; and criminal prosecution. The FDA also has the authority to request repair, replacement, or refund of the cost of any medical device manufactured or distributed by us. The FDA may also recommend prosecution to the U.S. Department of Justice. Any notice or communication from the FDA regarding a failure to comply with applicable requirements, or negative publicity or product liability claims resulting from any adverse regulatory action, could have a material adverse effect on our development of new laboratory tests, business strategy, financial condition, results of operations, or cash flows.
We have little control over the ongoing compliance of our suppliers with applicable regulations. Their failure to comply may expose us to regulatory action and other liability, including fines and civil penalties, suspension of production, suspension or delay in new product approval or clearance, product seizure or recall, or withdrawal of product approval or clearance.
Moreover, governmental authorities outside the U.S. have become increasingly stringent in their regulation of medical devices, and our products may become subject to more rigorous regulation by non-U.S. governmental authorities in the future. U.S. or non-U.S.
33
government regulations may be imposed in the future that may have a material adverse effect on our business and operations. The European Commission (“EC”) has harmonized national regulations for the control of medical devices through European Medical Device Directives with which manufacturers must comply. Under these new regulations, manufacturing plants must have received a full Quality Assurance Certification from a “Notified Body” in order to be able to sell products within the member states of the E.U. This Certification allows manufacturers to stamp the products of certified plants with a “CE” mark. Products covered by the EC regulations that do not bear the CE mark cannot be sold or distributed within the E.U. We have received certification for all currently existing manufacturing facilities.
In addition, until a completed mutual recognition agreement exists between Switzerland and the E.U., Switzerland will be considered a Third Country. The company has, however, pursued registration of certain key products in Switzerland under their new laws. Similar activities have been pursued in the United Kingdom in relation to Brexit.
Oversight of the medical device industry might affect the way we sell medical devices and compete in the marketplace.
The FDA, the U.S. Office of the Inspector General for the U.S. Department of Health and Human Services, the U.S. Department of Justice and other regulatory agencies actively enforce regulations prohibiting the promotion of a medical device for a use that has not been cleared or approved by the FDA. Use of a device outside its cleared or approved indications is known as “off-label” use. Physicians may prescribe our products for off-label uses, as the FDA does not restrict or regulate a physician’s choice of treatment within the practice of medicine. However, if a regulatory agency determines that our promotional materials, training or activities constitute improper promotion of an off-label use, the regulatory agency could request that we modify our promotional materials, training or activities, or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil fine and/or criminal penalties. Although our policy is to refrain from statements and activities that could be considered off-label promotion of our products, any regulatory agency could disagree and conclude that we have engaged in off-label promotion and, potentially, caused the submission of false claims. Moreover, the off-label use of our products may increase the risk of injury to patients, and, in turn, the risk of product liability claims. In addition, we may be subject to compliance actions, penalties, or injunctions if the FDA challenges one or more of our determinations that a product modification did not require new approval or clearance by the FDA.
An FDA panel recommended that bone growth stimulator devices be reclassified by the FDA from Class III to Class II devices, which could increase future competition for us in this product category and negatively affect our future sales of such products.
We have the market-leading bone growth stimulation platform with the only cervical spine indication granted by the FDA, and the only mobile device app accessory designed to help patients adhere to their prescriptions and improve their clinical outcomes, STIM onTrack 2.1. We also are investing in IDE studies to expand indications for use in areas such as rotator cuff tears. Our bone growth therapy products currently are designated as Class III devices. Class III devices are subject to the FDA’s most rigorous pathway to approval for medical devices in the U.S. The FDA may change classification of a device only if the proposed new class has sufficient regulatory controls to provide reasonable assurances of safety and effectiveness.
In September 2020, the FDA’s Orthopedic and Rehabilitation Devices Panel recommended that bone growth stimulator devices be reclassified from Class III to Class II devices with “special controls” to ensure patient safety and therapy efficacy. These proposed special controls include the condition that such devices be subject to rigorous clinical studies and post market surveillance for any new products. This would be in addition to other special controls and the Class II general requirement that any new products show “substantial equivalence” to already-cleared or approved devices.
We believe that the panel’s recommendation correctly recognizes the importance of PMA-like clinical data for these devices, so that manufacturers will continue to be required to submit robust clinical data under the approval or clearance process to ensure the safety and efficacy of these devices for patients. We, along with other bone growth stimulation manufacturers, submitted comments in response to the FDA’s proposed rulemaking to underscore the panel’s recommendation of the need for robust clinical data prior to approval or clearance of bone growth stimulator products, together with post market surveillance requirements.
In the long-term, the recommended reclassification could enhance the ability of competitors to enter the market if they are able to create technologies with comparable efficacy to our devices, which could result in our products facing additional competition, thereby negatively affecting our future sales of these products.
We continue to be affected by U.S. healthcare reform initiatives.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (or collectively the “ACA”), has caused a number of substantial changes to occur in recent years in the way healthcare is financed by both governmental
34
and private insurers. The ACA is far-reaching and is intended to expand access to health insurance coverage, improve quality, and reduce costs over time. Among other things, the ACA:
Established a Patient-Centered Outcomes Research Institute to oversee and identify priorities in comparative clinical effectiveness research in an effort to coordinate and develop such research; and
Implemented payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians, and other providers to improve the coordination, quality, and efficiency of certain healthcare services through bundled payment models.
U.S. government agencies continue efforts to modify provisions of the ACA. For example, CMS began permitting states to impose work requirements on persons covered by Medicaid expansion plans, certain federal subsidies to insurers have ended, and certain short-term insurance plans not offering the full array of ACA benefits have been allowed to extend in duration. Some of these changes are being challenged in U.S. courts and so their long-term impact remains uncertain. This changing federal landscape has both positive and negative impacts on the U.S. healthcare industry, with much remaining uncertain as to how various provisions of federal law, and potential modification or repeal of these laws, will ultimately affect the industry. Persisting uncertainty with respect to the scope and effect of certain provisions of the ACA have made compliance costly. Any future changes to the ACA or other such legislation, depending on their nature, could affect rebates, prices, or the rate of price increases for health care products and services, or required reporting and disclosure, and could have an adverse effect on our ability to maintain or increase sales of any of our products and achieve profitability. We cannot predict the timing or impact of any future rulemaking or changes in the law. However, any changes that have the effect of reducing reimbursements for our products or reducing medical procedure volumes could have a material and adverse effect on our business, financial condition, and results of operations.
We are subject to differing customs and import/export rules in several jurisdictions in which we operate.
We import and export our products to and from a number of different countries around the world. Foreign governmental regulations have become increasingly stringent and more common, and we may become subject to even more rigorous regulation by foreign governmental authorities. Numerous laws restrict, and in some cases prohibit, U.S. companies from directly or indirectly selling goods, technology or services to people or entities in certain countries. In addition, these laws require that we exercise care in structuring our sales and marketing practices and effecting product registrations in foreign countries. Compliance with these regulations is costly.
The import and export of our products involve subsidiaries and third parties operating in jurisdictions with different customs and import/export rules and regulations. Customs authorities in such jurisdictions may challenge our treatment of customs and import/export rules relating to product shipments under aspects of their respective customs laws and treaties. If we are unsuccessful in defending our treatment of customs and import/export classifications, we may be subject to additional customs duties, fines, or penalties that could adversely affect our profitability.
In addition, changes in U.S. or foreign policies regarding international trade could also negatively impact our business. The enactment of or increases in tariffs, or other such charges, on specific products that we sell or with which our products compete, may have an adverse effect on our business or on our results of operations.
The sales and marketing practices of our industry have been the subject of increased scrutiny from federal and state government agencies.
AdvaMed (U.S.), EucoMed (Europe), MEDEC (Canada) and MTAA (Australia), some of the principal trade associations for the medical device industry, have promulgated model codes of ethics that set forth standards by which its members should (and non-member companies may) abide in the promotion of their products in various regions. We have implemented policies and procedures for compliance consistent with those promulgated by these associations, and we train our sales and marketing personnel on our policies regarding sales and marketing practices. Nevertheless, the sales and marketing practices of our industry have been the subject of increased scrutiny from federal and state government agencies, we believe this trend will continue and that it could affect our ability to retain customers and other relationships important to our business.
For example, prosecutorial scrutiny and governmental oversight, at both the state and federal levels, over some major device companies regarding the retention of healthcare professionals have limited how medical device companies may retain healthcare professionals as consultants. Various hospital organizations, medical societies and trade associations are establishing their own practices that may require detailed disclosures of relationships between healthcare professionals and medical device companies or ban or restrict certain marketing and sales practices, such as gifts and business meals. In addition, the ACA, as well as certain state laws, require detailed disclosure of certain financial relationships, gifts and other remuneration made to certain healthcare professionals and teaching hospitals, the publicity surrounding which could have a negative impact on our relationships with our
35
customers and ability to seek input on product design or involvement in research. As a result of laws, rules and regulations or our own or third-party policies that prohibit or restrict interactions, or the growing perception that any interaction between healthcare professionals and industry are tainted, we may be unable to engage with our healthcare professional customers in the same manner or to the same degree, or at all, as would otherwise be the case, which may adversely affect our ability to understand our customer’s needs and to incorporate into our development programs feedback that addresses these needs. If we are unable to develop and commercialize new products that address the needs of our physician customers and their patients, our products may not be broadly accepted in the marketplace, or at all, which would have a negative effect on our business, results of operations and financial condition.
We are subject to requirements relating to hazardous materials which may impose significant compliance or other costs on us.
Our research, development and manufacturing processes involve the controlled use of certain hazardous materials. For example, our allograft bone tissue processing may generate waste materials that in the U.S. are classified as medical waste. In addition, we lease facilities at which hazardous materials could have been used. Because of the foregoing, we are subject to federal, state, foreign and local laws and regulations governing the use, manufacture, storage, handling, treatment, remediation and disposal of hazardous materials and certain waste products.
Although we believe that our procedures for handling and disposing of hazardous materials comply with applicable laws as currently in effect, we cannot eliminate the risk of accidental contamination or injury from these materials. In addition, under some environmental laws and regulations, we could also be held responsible for all of the costs relating to any contamination at our past or present facilities and at third-party waste disposal sites, even if such contamination was not caused by us. If an accident occurs, state or federal or other applicable authorities may curtail our use of these materials and interrupt our business operations. In addition, if an accident or environmental discharge occurs, or if we discover contamination caused by prior operations, including by prior owners and operators of properties we acquire, we could be liable for cleanup obligations, damages and fines any related liability could exceed our resources. If such unexpected costs are substantial, this could significantly harm our financial condition and results of operations. We carry no insurance specifically covering environmental claims relating to the use of hazardous materials.
Risks Related to our Business and Industry
The COVID-19 pandemic has materially adversely affected, and could continue to materially adversely affect, our operations, supply chain, manufacturing, product demand, product distribution, customers and other business activities.
The novel coronavirus discovered in late 2019, and the disease it causes, known as COVID-19, has led to significant disruptions in the healthcare market and the United States and international economies that may continue for a prolonged duration. The rapid spread of the coronavirus in 2020 and variants of the virus in 2021, the persistence of the resulting pandemic, the measures governments and private parties have implemented in order to stem the spread of this pandemic, and the general concern about the virus, have had, and could continue to have, a negative effect on the demand for many of our products compared to historical levels, and consequently upon our business. In particular, many of our products are particularly sensitive to reductions in elective medical procedures. Elective medical procedures were suspended or reduced at various times in 2020, 2021, and portions of 2022, in many of the markets where our products are marketed and sold, which negatively affected our business, cash flows, financial condition and results of operations.
Deferrals of elective surgeries could result in delayed product launches if it takes longer than anticipated to collect feedback following an alpha launch. Further, facilities at which our products typically are used may not reopen or, even if they reopen, patients may elect to have procedures performed at facilities that are, or are perceived to be, lower-risk, such as ambulatory surgery centers, and our products may not be approved at such facilities, and we may be unable to have our products approved for use at such facilities on a timely basis, or at all.
The future trajectory of the COVID-19 pandemic remains uncertain, both in the U.S. and in other markets, particularly due to the uncertainty as to the nature of future variants, and whether vaccines will protect against severe illness with respect to such future variants.
Given these various uncertainties, it is unclear the extent to which lingering slowdowns in elective procedures will continue to affect our business in 2023 and beyond. We expect that the effects of COVID-19 on our business will depend on various factors including (i) the magnitude, length and virulence of additional case waves and future variants, (ii) the continued distribution, efficacy, refinement, and public acceptance of COVID-19 vaccines, (iii) the comfort level of patients in visiting clinics and hospitals, and (iv) the extent to which further elective surgery slowdowns occur during periods when hospital capacity is stretched because of the need to treat COVID-19 patients.
36
In addition to its effect on elective surgeries, the pandemic could also negatively affect our ability, and the ability of our third-party suppliers, manufacturers, distributors, and customers, to retain key employees and ensure the continued service and availability of skilled personnel necessary to run our, and their, complex operations. To the extent our management or other personnel, or the management or other personnel of our third-party suppliers, manufacturers, distributors, and customers, are negatively affected by the pandemic and are not available to perform their job duties, we could experience delays in, or the suspension of, our manufacturing operations, sales activities, research and product development activities, regulatory work streams, clinical development programs and other important commercial and corporate functions. Moreover, our relationships with our employees may be disrupted due to measures implemented in response to the COVID-19 pandemic. We have observed an overall tightening and increasingly competitive labor market due to labor shortages caused in part by the COVID-19 pandemic and responsive measures, which has included increased wages offered by other employers and voluntary attrition of employees in the industry, including at third-party suppliers, manufacturers, distributors and customers.
All of these factors, collectively, could materially adversely affect our business, financial condition and results of operations.
The COVID-19 pandemic and related supply chain and raw material disruptions, and the ongoing conflict between Russia and Ukraine, and the global response to it, could have a continuing material impact on our global operations and the operations of our supply chain, which could adversely impact our business results and financial condition.
We rely on a limited number of suppliers to manufacture or supply certain products or components. In the event of interruption within our supply chain, or global shortages of key supplies or components, we may not be able to increase capacity from other sources or develop alternative or secondary sources without incurring significant additional costs and/or substantial delays. For example, the COVID-19 pandemic has led to a global shortage of semiconductor chips, which are used in certain of our products. This shortage appears primarily to have been caused by manufacturers experiencing shutdowns or slowdowns during the pandemic, and it may take several fiscal quarters or longer for normalized capacity to return. In addition, limitations in key raw material supplies could also cause semiconductor chip and other component shortages to continue. To the extent it continues, or more shortages are experienced, particularly on a longer term basis, this could adversely affect our ability to procure such components and manufacture certain of our products or it could require us to redesign any affected products in order to incorporate more readily available components, which may require additional regulatory testing and approvals. Thus, our business could be adversely affected in a significant manner if one or more of our suppliers are impacted by any interruption at a particular location or in relation to a particular material or component.
The ongoing conflict between Russia and Ukraine has resulted in the implementation of sanctions by the United States and other governments against Russia and has caused significant volatility and disruptions to the global markets. It is not possible to predict the short- or long-term implications of this conflict, which could include but are not limited to further sanctions, uncertainty about economic and political stability, increases in inflation rate and energy prices, supply chain challenges and adverse effects on currency exchange rates and financial markets. In addition, the United States government reported that United States sanctions against Russia in response to the conflict could lead to an increased threat of cyberattacks against United States companies. These increased threats could pose risks to the security of our information technology systems and networks, as well as the confidentiality, availability and integrity of our data. A significant escalation or further expansion of the conflict's current scope or related disruptions to the global markets could have a material adverse effect on our results of operations.
Our business may be adversely affected if consolidation in the healthcare industry leads to demand for price concessions or if a group purchasing organization (“GPO”) or similar entity excludes us from being a supplier.
Because healthcare costs have risen significantly over the past decade, numerous initiatives and reforms have been launched by legislators, regulators, and third-party payors to curb these costs. As a result, there has been a trend toward healthcare cost containment through aggregating purchasing decisions and industry consolidation, along with the growth of managed care organizations, all of which has placed increased emphasis on the delivery of more cost-effective medical therapies. For example:
•There has been consolidation among healthcare facilities and purchasers of medical devices, particularly in the U.S. One of the results of such consolidation is that GPOs, integrated delivery networks and large single accounts use their market power to consolidate purchasing decisions, which intensifies competition to provide products and services to healthcare providers and other industry participants, resulting in greater pricing pressures and the exclusion of certain suppliers from important market segments. For example, some GPOs negotiate pricing for its member hospitals and require us to discount, or limit our ability to increase, prices for certain of our products. In particular, certain of our demineralized bone matrix (“DBM”) products are priced at a premium to competitors' DBM products and a significant price reduction could result in a material adverse effect on our profitability.
37
•Physicians increasingly have moved from independent, out-patient practice settings toward employment by hospitals and other larger healthcare organizations, which align physicians’ product choices with their employers’ price sensitivities and adds to pricing pressures. Hospitals have introduced and may continue to introduce new pricing structures into their contracts to contain healthcare costs, including fixed price formulas and capitated and construct pricing.
•Certain hospitals provide financial incentives to doctors for reducing hospital costs (known as gainsharing), rewarding physician efficiency (known as physician profiling) and encouraging partnerships with healthcare service and goods providers to reduce prices.
•Existing and proposed laws, regulations, and industry policies, in both domestic and international markets, regulate or seek to increase regulation of sales and marketing practices and the pricing and profitability of companies in the healthcare industry.
As the healthcare industry consolidates, competition to provide products and services to industry participants has become and may continue to become more intense. This has resulted and may continue to result in greater pricing pressures and the exclusion of certain suppliers from important markets as GPOs, independent delivery networks, and large single accounts continue to use their market power to consolidate purchasing decisions and as larger manufacturers use their broad offerings to secure exclusive arrangements. If a GPO were to exclude us from their supplier list, our net sales could be adversely impacted. We expect that market demand, government regulation, third-party reimbursement policies, and societal pressures will continue to change the worldwide healthcare industry, which may exert further downward pressure on the prices of our products.
In addition, the largest device companies with multiple product franchises have increased their effort to leverage and contract broadly with customers across franchises by providing volume discounts and multi-year arrangements that could prevent our access to these customers or make it difficult (or impossible) to compete on price.
The industry in which we operate is highly competitive. New developments by others could make our products or technologies non-competitive or obsolete.
The medical devices industry is highly competitive. We compete with a large number of companies, many of which have significantly greater financial, manufacturing, marketing, distribution, and technical resources than we do. Many of our competitors may be able to develop products and processes competitive with, or superior to, our own. Our competitors may also have: stronger intellectual property portfolios; broader spine surgery product offerings and products supported by more extensive clinical data; more established distribution networks; entrenched relationships with physicians; significantly greater name recognition and more recognizable trademarks for products similar to the products we sell; more established relationships with healthcare providers and payors; greater experience in obtaining and maintaining FDA and other regulatory clearances or approvals for products and product enhancement; and greater experience in launching, marketing and selling products than we do. Many of our competitors specialize in a specific product or focus on a particular market segment, making it more difficult for us to increase our overall market position. The frequent introduction by competitors of products that are, or claim to be, superior to our products, or that are alternatives to our existing or planned products may also create market confusion that may make it difficult to differentiate the benefits of our products over competing products. In addition, the entry of multiple new products and competitors may lead some of our competitors to employ pricing strategies that could adversely affect the pricing of our products and pricing in the spine market generally.
Furthermore, we may not be able to successfully develop or introduce new products that are less costly or offer better performance than those of our competitors, or offer purchasers of our products payment and other commercial terms as favorable as those offered by our competitors. For more information regarding our competitors, see Item 1, “Business,” under the subheading “Competition.”
In addition, the spine and orthopedic medical device industry in which we compete is undergoing, and is characterized by, rapid and significant technological change. We expect competition to intensify as technological advances are made. New technologies and products developed by other companies are regularly introduced into the market, which may render our products or technologies non-competitive or obsolete.
Our ability to market products successfully depends, in part, upon the acceptance of the products not only by consumers, but also by independent third parties.
Our ability to market our products successfully depends, in part, on the acceptance of the products by independent third parties (including hospitals, physicians, other healthcare providers, and third-party payors) as well as patients. Market acceptance for any of our products requires, among other things, that we timely secure regulatory clearance and/or approval; demonstrate the value of
38
our products, both to our physician customers and payors, which may require that we collect clinical data and/or conduct clinical studies; effectively educate and train our physician customers and their staff on the proper use of our products; obtain and maintain coverage and adequate reimbursement for our products, both within and outside the U.S., including under Medicare and Medicaid and from private payors; attract and retain a network of independent sales agents and stocking distributors focused on neurophysicians and orthopedic spine physicians; develop and execute an effective marketing strategy; protect the proprietary positions of our products, including through patent protection; and consistently produce quality products in sufficient quantities to meet demand. Significant risks are associated with each of these activities and other activities required to achieve market acceptance of both our current and future products, including risks inherent in collaborations, or use of nascent manufacturing or imaging techniques, such as additive processing (more commonly known as 3D printing) or advanced optical technologies and machine version-based registration algorithms. Unanticipated side effects or unfavorable publicity concerning any of our products could have an adverse effect on our ability to maintain hospital approvals or achieve acceptance by prescribing physicians, managed care providers and other retailers, customers, and patients.
Clinical studies are expensive and subject to extensive regulation and their results may not support our product candidate claims or may result in the discovery of adverse effects.
In developing new products or new indications for, or modifications to, existing products, we may conduct or sponsor pre-clinical testing, clinical studies or other clinical research. We are conducting post-market clinical studies of some of our products to gather information about their performance or optimal use. The data collected from these clinical studies may ultimately be used to support additional market clearance or approval for these products or future products. If any of our new products require premarket clinical studies, these studies are expensive, the outcomes are inherently uncertain and they are subject to extensive regulation and review by numerous governmental authorities both in the U.S. and abroad, including by the FDA and, if federal funds are involved or if an investigator or site has signed a federal assurance, are subject to further regulation by the Office for Human Research Protections and the National Institutes of Health. For example, clinical studies must be conducted in compliance with FDA regulations, local regulations, and according to principles and standards collectively called “Good Clinical Practices.” Failure to comply with applicable regulations could result in regulatory and legal enforcement action, including fines, penalties, suspension of studies, and also could invalidate the data and make it unusable to support an FDA submission.
Even if any of our future premarket clinical studies are completed as planned, we cannot be certain that their results will support our product candidates and/or proposed claims or that the FDA or foreign authorities and Notified Bodies will agree with our interpretation and conclusions regarding the data they generate. Success in pre-clinical studies and early clinical studies does not ensure that later clinical studies will succeed, and we cannot be sure that the results of later studies will replicate those of earlier or prior studies. The clinical study process may fail to demonstrate that our product candidates are safe and effective for the proposed indicated uses, which could cause us to abandon a product candidate and may delay development of others. Any delay or termination of our clinical studies will delay the filing of our product submissions and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patient subjects enrolled in our clinical studies of our marketed products will experience adverse side effects that are not currently part of the product candidate’s profile and, if so, these findings may result in lower market acceptance, which could have a material and adverse effect on our business, results of operations and financial condition.
Further, the COVID-19 pandemic could limit or restrict our ability or the ability of others on which we rely to initiate, conduct, or continue our clinical studies of some of our products. Delays and disruption in such studies could result in delays for expanded FDA and other regulatory clearance or approval of our products.
If the third parties on which we rely to conduct our clinical studies and to assist us with pre-clinical development do not perform as contractually required or expected, we may not obtain regulatory clearance, approval or a CE Certificate of Conformity for or commercialize our products.
We often must rely on third parties, such as contract research organizations, medical institutions, clinical investigators, and contract laboratories, to assist in conducting our clinical studies and other development activities. If these third parties do not successfully carry out their contractual duties, comply with applicable regulatory obligations or meet expected deadlines, or if these third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to failing to adhere to clinical protocols, to applicable regulatory requirements or otherwise, our pre-clinical development activities and clinical studies may be extended, delayed, suspended or terminated. Under these circumstances, we may not be able to obtain regulatory clearance/approval or a CE Certificate of Conformity for, or successfully commercialize, our products on a timely basis, if at all, and our business, operating results and prospects may be materially and adversely affected.
39
Our allograft and cellular bone allografts could expose us to certain risks that could disrupt our business.
Our Biologics business markets allograft tissues that are derived from human cadaveric donors, and our ability to market the tissues depends on our supplier continuing to have access to donated human cadaveric tissue, as well as the maintenance of high standards by the supplier in its processing methodology. The supply of such donors is inherently unpredictable and can fluctuate over time. The allograft tissues are regulated under the FDA’s HCT/P regulatory paradigm and not as a medical device, biologic, or drug. There can be no assurance that the FDA will not at some future date re-classify the allograft tissues, and the reclassification of this product from a human tissue to a medical device could have adverse consequences for us or for the supplier of this product and make it more difficult or expensive for us to conduct this business by requiring premarket clearance or approval, as well as compliance with additional post-market regulatory requirements.
In addition, procurement of certain human organs and tissue for transplantation is subject to the National Organ Transplant Act (the “NOTA”), which prohibits the transfer of certain human organs, including skin and related tissue, for valuable consideration, but permits the reasonable payment associated with the removal, transportation, implantation, processing, preservation, quality control and storage of human tissue and skin. If we were to be found to have violated NOTA’s prohibition on the sale or transfer of human tissue for valuable consideration, we would potentially be subject to criminal enforcement sanctions, which could materially and adversely affect our results of operations.
Because of the absence of a harmonized regulatory framework and the proposed regulation for advanced therapy medicinal products in the E.U., as well as for other countries, the approval process in the E.U. for human-derived cell or tissue-based medical products could be extensive, lengthy, expensive and unpredictable. Among others, some of our Biologics products are subject to E.U. member states’ regulations that govern the donation, procurement, testing, coding, traceability, processing, preservation, storage and distribution of HCT/Ps. These E.U. member states’ regulations include requirements for registration, listing, labeling, adverse-event reporting and inspection and enforcement. Some E.U. member states have their own tissue banking regulations, including new requirements related to COVID-19 and donor screening. Non-compliance with various regulations governing our products in any E.U. member state could result in the banning of our products in such member state or enforcement actions being brought against us, which could have a material and adverse effect on our business, results of operations and financial condition.
Unfavorable media reports or other negative publicity concerning both alleged improper methods of tissue recovery from donors and disease transmission from donated tissue could limit widespread acceptance of some of our products.
Unfavorable reports of improper or illegal tissue recovery practices, both in the U.S. and internationally, as well as incidents of improperly processed tissue leading to the transmission of disease, may affect the rate of future tissue donation and market acceptance of technologies incorporating human tissue. In addition, negative publicity could cause the families of potential donors to become reluctant to donate tissue to for-profit tissue processors. For example, the media has reported examples of alleged illegal harvesting of body parts from cadavers and resulting recalls conducted by certain companies selling human tissue-based products affected by the alleged illegal harvesting. These reports and others could have a negative effect on our tissue regeneration business.
Certain of our products contain materials derived from animal sources and may become subject to additional regulation.
Certain of our products contain material derived from bovine tissue. Products that contain materials derived from animal sources, including food, pharmaceuticals and medical devices, are subject to scrutiny in the media and by regulatory authorities. Regulatory authorities are concerned about the potential for the transmission of disease from animals to humans via those materials. In past years, public scrutiny was particularly acute in Western Europe with respect to products derived from animal sources, largely due to concern that materials infected with the agent that causes BSE otherwise known as mad cow disease, may, if ingested or implanted, cause a variant of the human Creutzfeldt-Jakob disease, an ultimately fatal disease with no known cure. Cases of BSE in cattle discovered in Canada and the U.S. increased awareness in North America.
Products that contain materials derived from animals, including our products, could become subject to additional regulation, or even be banned in certain countries, because of concern over the potential for the transmission of infectious or other agents. Significant new regulation, or a ban of our products, could have a material and adverse effect on our business or our ability to expand our business.
Certain countries, such as Japan, China, Taiwan, and Argentina, have issued regulations that require our collagen products be processed from bovine tendon sourced from countries where no cases of BSE have occurred. The collagen raw material we use in our products is sourced from New Zealand. Our supplier has obtained approval from certain countries, including the U.S., the E.U., Japan, Taiwan, China, and Argentina, for the use of such collagen raw material in products sold in those countries. If we cannot continue to obtain collagen raw material from a qualified source of tendon from a country that has never had a case of BSE, we will
40
not be permitted to sell our collagen products in certain countries, which could have a material and adverse effect on our business, results of operations and financial condition.
We may not be able to successfully introduce new products to the market and market opportunities that we expect to develop for our products may not be as large as we expect.
To be and remain competitive, we need to continue to make improvements in our products, develop new products, introduce our products into new markets, and successfully respond to technological advances. Doing so is technologically challenging and involves significant risks and uncertainty. Despite our planning, the process of developing and introducing new products (including product enhancements) is inherently complex and uncertain, and involves risks. The success of any of our new product offerings or enhancement or modification to our existing products will depend on several factors, including our ability to:
•properly identify and anticipate physician and patient needs;
•develop new products or enhancements or modifications in a timely manner;
•obtain regulatory clearance and/or approvals for new products or product enhancements or modifications in a timely manner;
•achieve timely alpha and/or full commercial launches of new products;
•provide adequate training to potential users of new products and product enhancements or modifications;
•receive adequate reimbursement approval of third-party payors such as Medicaid, Medicare and private insurers;
•gain broad market acceptance (including by physicians);
•and
develop an effective marketing and distribution network.
In addition, competitors could develop products that are more effective, are less expensive to manufacture, are priced more competitively or are ready for commercial introduction before our products. The introduction of new products by our competitors may lead us to reduce the prices of our products, may lead to reduced margins or loss of market share, and may render our products obsolete or noncompetitive.
These risks make it inherently difficult to forecast and predict the future net sales of our products. If we cannot develop technically and commercially viable new products and enhancements or modifications to our existing products on a consistent basis and before our competitors, our prospects could be materially and adversely affected. In addition, if the market opportunities that we expect to develop for our products, including new products, are not as large as we expect, it could adversely affect our ability to grow our business.
It is also important that we carefully manage our introduction of new products and enhancements or modifications to our existing products. If potential customers delay purchases until new or enhanced or modified products are available, it could negatively impact our sales. In addition, to the extent we have excess or obsolete inventory as we transition to new or enhanced or modified products, it would result in margin reducing write-offs for obsolete inventory, and our results of operations may suffer.
There is no guarantee that the FDA will grant 510(k) clearance or premarket approval, or that equivalent foreign regulatory authorities will grant the foreign equivalent, of our future products, and failure to obtain necessary clearances or approvals for our future products would adversely affect our ability to grow our business.
In general, unless an exemption applies, a medical device and modifications to the device or its indications must receive either premarket approval or premarket clearance from the FDA before it can be marketed in the U.S. While in the past we have received such clearances, we may not succeed in the future in receiving approvals and clearances in a timely manner, or at all. The process of obtaining approval or clearance from the FDA and comparable foreign regulatory agencies for new products, or for enhancements or modifications to existing products, could:
•require the expenditure of substantial resources;
41
•involve rigorous and expensive pre-clinical and clinical testing, as well as post-market surveillance;
•involve modifications, repairs, or replacements of our products; and
•result in limitations on the indicated uses of our products.
Some of our new products will require FDA 510(k) clearance or approval of a premarket approval application, or PMA, prior to being marketed. Any modification to a 510(k)-cleared device that could significantly affect its safety or effectiveness, including significant design and manufacturing changes, or that would constitute a major change in its intended use, design or manufacture, requires a new 510(k) clearance or, possibly, approval of a PMA. Similarly, modifications to PMA-approved products may require submission and approval of a PMA supplement. The FDA requires every manufacturer to determine whether a new 510(k) or PMA is needed in the first instance, and the FDA has issued guidance on assessing modifications to 510(k)-cleared and PMA-approved devices to assist manufacturers with making these determinations. However, the FDA may review any such determination and the FDA may not agree with our determinations regarding whether new clearances or approvals are necessary. We have modified some of our 510(k)-cleared products and have determined, based on our understanding of FDA guidance, that certain changes did not require new 510(k) clearances. If the FDA disagrees with our determination and requires us to seek new 510(k) clearances, or PMA approval, for modifications to our cleared products, we may have to stop marketing or distributing our products, we may need to recall the modified product until we obtain clearance or approval, and we may be subject to significant regulatory fines or penalties. Significant delays in receiving clearance or approval, or failing to receive clearance or approval for our new products would have a material and adverse effect on our ability to expand our business.
Outside the U.S., clearance or approval procedures can vary among countries and can involve additional product testing and validation and additional administrative review periods. The time required to obtain clearance or approval in other countries might differ from that required to obtain FDA clearance or approval. The regulatory process in other countries may include all of the risks to which we are exposed in the U.S., as well as other risks. Favorable regulatory action in one country does not ensure favorable regulatory action in another, but a failure or delay in obtaining regulatory clearance or approval in one country may have a negative effect on the regulatory process in others. Failure to obtain clearance or approval in other countries or any delay or setback in obtaining such clearance or approval have a material and adverse effect on our business, including that our products may not be cleared or approved for all indications requested, which could limit the uses of our products and have an adverse effect on product sales.
In the European Economic Area (“EEA”), we must inform the Notified Body that carried out the conformity assessment of the medical devices we market or sell in the EEA of any planned substantial change to our quality system or any significant change to our devices. The Notified Body will then assess the change and verify whether it affects the products’ conformity with the Essential Requirements or the conditions for the use of the device. If the assessment is favorable, the Notified Body may issue a new CE Certificate of Conformity or an addendum to the existing CE Certificate of Conformity. If it is not, we may not be able to continue to market and sell the applicable product in the EEA, which could have a material and adverse effect on our business, results of operations and financial condition.
We cannot be certain that we will receive required approval or clearance from the FDA and foreign regulatory agencies for new products, including modifications to existing products, on a timely basis, or at all. Failing to receive approval or clearance for new products on a timely basis would have a material and adverse effect on our financial condition and results of operations.
Growing our business requires that we properly educate and train physicians regarding the distinctive characteristics, benefits, safety, clinical efficacy, and cost-effectiveness of our products.
Acceptance of our products depends in part on our ability to (i) educate the medical community as to the distinctive characteristics, benefits, safety, clinical efficacy, and cost-effectiveness of our products compared to alternative products, procedures, and therapies, and (ii) train physicians in the proper use and implementation of our products. This is particularly true in instances of newly launched products or in the introduction of a product into a new market, such as our launch of the M6-C artificial cervical disc within the U.S. We support our sales force and distributors through specialized training workshops in which physicians and sales specialists participate. We also produce marketing materials, including materials outlining surgical procedures, for our sales force and distributors in a variety of languages using printed, video, and multimedia formats. To provide additional advanced training for physicians, consistent with the AdvaMed Code and the MedTech Code, we organize regular multilingual teaching seminars in multiple locations. However, convincing physicians to dedicate the time and energy necessary for adequate training is challenging, and we may not be successful in our efforts to educate the medical community and properly train physicians. Physicians who do not use our products may be hesitant to do so for the following or other reasons:
42
•lack of experience with our products, techniques, or technologies, or with the equipment necessary to use any of the foregoing;
•existing relationships with those who sell competitive products;
•the time required for physician and medical staff education and training on new products, techniques and equipment and technologies;
•lack or perceived lack of clinical evidence supporting patient benefit relative to competing products;
•our products not being included on hospital formularies, in integrated delivery networks or on group purchasing organization preferred vendor lists;
•less attractive coverage and/or reimbursement within healthcare payment systems for our products and procedures compared to other products and procedures;
•other costs associated with introducing new products and the equipment necessary to use new products; and
•perceived risk of liability that could be associated with the use of new products, techniques, or technologies.
If physicians are not properly trained, they may misuse or ineffectively use our products, which may result in unsatisfactory patient outcomes, patient injury, negative publicity, or lawsuits against us. In addition, a failure to educate the medical community regarding our products may impair our ability to achieve market acceptance of our products.
In addition, we believe recommendations and support of our products by influential physicians are essential for market acceptance and adoption. If we do not receive support from such physicians or long-term data does not show the benefits of using our products, physicians may not use our products. If we are not successful in convincing physicians of the merits of our products, we may not maintain or grow our sales or achieve or sustain profitability.
Relatedly, although we believe our training methods for physicians are conducted in compliance with FDA and other applicable regulations developed both nationally and in third countries, if the FDA or other regulatory agency determines that our training constitutes promotion of an unapproved use or promotion of an intended purpose not covered by the CE mark affixed to our products or FDA approved labeling, they could request that we modify our training or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil fine and criminal penalty.
Sales of, or the price at which we sell, our products may be adversely affected unless the safety and efficacy of our products, alone and relative to competitive products, is demonstrated in clinical studies.
Generally, we have obtained 510(k) clearance to manufacture, market and sell the products we market in the U.S. and the right to affix the CE mark to the products we market in the EEA. To date, we have not been required to generate new clinical data to support our 510(k) clearances, CE marks, or product registrations in other countries. However, the EU Medical Device Regulations, which replaced the prior medical device directives in May 2021, require submission of certain pre- and post-market data to maintain our CE marks. Additionally, we recently completed an analysis of which of our product systems will require submission of clinical data pursuant to MEDDEV 2.7.1 rev 4, which sets forth the European Commission’s guidance on the clinical evaluation of medical devices. Accordingly, and in line with our vision to deliver clinical value, we have commenced clinical data collection activities for certain of our marketed products as more fully described elsewhere in this "Risk Factors" section.
In part due to the increased emphasis on the delivery of more cost-effective treatments, purchasing decisions of our customers increasingly will be based on clinical data that demonstrates the value of our products or the effectiveness of our products relative to others. Conducting clinical studies is expensive and time-consuming and outcomes are uncertain. See “Clinical studies are expensive and subject to extensive regulation and their results may not support our product candidate claims or may result in the discovery of adverse effects,” above. We may elect not to, or may be unable to, fund the clinical studies necessary to generate the data required for all of our products to compete effectively, in part due to the breadth of our product portfolio. Currently, we do not expect to undertake such clinical studies for all of our products and only expect to do so where we anticipate the benefits will outweigh the costs on a risk-adjusted basis. However, even when we elect and are able to fund such clinical studies on one or more of our products, such studies may not succeed. Data we generate may not be consistent with our existing data and may demonstrate less favorable safety or efficacy, which could reduce demand for our products and negatively impact future sales. Neurophysicians and orthopedic spine physicians may be less likely to use our products if more robust, or any, clinical data supporting the safety and efficacy of competing products is available. If we are unable to or unwilling to generate clinical data supporting the safety and effectiveness of our products, our business, results of operations and financial condition could be materially and adversely affected.
43
Further, future patient studies or clinical experience may indicate that treatment with our products does not improve patient outcomes.
With the passage of the American Recovery and Reinvestment Act of 2009, funds have been appropriated for the U.S. Department of Health and Human Services’ Healthcare Research and Quality to conduct comparative effectiveness research to determine the effectiveness of different drugs, medical devices, and procedures in treating certain conditions and diseases. Some of our products or procedures performed with our products could become the subject of such research. It is unknown what effect, if any, this research may have on our business. Further, future research or experience may indicate that treatment with our products does not improve patient outcomes or improves patient outcomes less than we initially expected. Such results would reduce demand for our products, affect sustainable reimbursement from third-party payers, significantly reduce our ability to achieve expected revenue, and could cause us to withdraw our products from the market and could prevent us from sustaining or increasing profitability. Moreover, if future results and experience indicate that our products cause unexpected or serious complications or other unforeseen negative effects, we could be subject to significant legal liability, negative publicity, and damage to our reputation, and we could experience a dramatic reduction in sales of our products, all of which would have a material adverse effect on our business, financial condition, and results of operations. The spine medical device market has been particularly prone to potential product liability claims that are inherent in the testing, manufacture, and sale of medical devices and products for spine surgery procedures.
We may be adversely affected by any disruption in our information technology systems, which could adversely affect our cash flows, operating results, and financial condition.
Our operations are dependent upon our information technology systems, which encompass all of our major business functions. We rely upon such information technology systems to manage and replenish inventory, fill and ship customer orders on a timely basis, coordinate our sales activities across all of our products and services, and coordinate our administrative activities. A substantial disruption in our information technology systems for any prolonged time period (arising from, for example, system capacity limits from unexpected increases in our volume of business, outages, or delays in service) could result in delays in receiving inventory and supplies or filling customer orders and adversely affect our customer service and relationships. Our systems might be damaged or interrupted by natural or man-made events, or by computer viruses, physical or electronic break-ins, and similar disruptions affecting the internet. There can be no assurance that such delays, problems, or costs will not have a material adverse effect on our cash flows, operating results, and financial condition.
As our operations grow in both size and scope, we will continuously need to improve and upgrade our information technology systems and infrastructure while maintaining the reliability and integrity of our information technology systems and infrastructure. An expansion of our information technology systems and infrastructure may require us to commit substantial financial, operational, and technical resources before the volume of our business increases, with no assurance that the volume of business will increase. Any such upgrades to our information technology systems and information technology, or new technology, now and in the future, require that our management and resources be diverted from our core business to assist in integrating such upgrades or new technology. There can be no assurance that the time and resources our management will need to devote to these upgrades, service outages, or delays due to the installation of any new or upgraded technology (and customer issues therewith), or the impact on the reliability of our data from any new or upgraded technology, will not have a material adverse effect on our cash flows, operating results, and financial condition.
A significant portion of our operations run on a single Enterprise Resource Planning (“ERP”) platform. To manage our international operations efficiently and effectively, we rely heavily on our ERP system, internal electronic information and communications systems, and on systems or support services from third parties. Any of these systems are subject to electrical or telecommunications outages, computer hacking, or other general system failure. It is also possible that future acquisitions will operate on different ERP systems and that we could face difficulties in integrating operational and accounting functions of new acquisitions. Difficulties in upgrading or expanding our ERP system or system-wide or local failures that affect our information processing could adversely affect our cash flows, operating results, and financial condition.
We may be adversely affected by a failure or compromise from a cyber-attack, data breach or ransomware attack, which could have an adverse effect on our business.
We rely on information technology systems to perform our business operations, including processing, transmitting, and storing electronic information, and interacting with customers, suppliers, healthcare payors, and other third parties. Like other medical device companies, the size and complexity of our information technology systems make them vulnerable to a cyber-attack, malicious intrusion, breakdown, destruction, loss of data privacy, ransomware attack, or other significant disruption. Our information systems
44
require an ongoing commitment of significant resources to maintain, protect, and enhance existing systems and develop new systems to keep pace with continuing changes in information processing technology, evolving systems and regulatory standards, the increasing need to protect financial or personal information related to patients and customers, and changing customer patterns.
For example, third parties may attempt to hack into our products to obtain data relating to patients, disrupt the performance of our products, or access our proprietary information. We could also be subject to a ransomware attack, which is a type of malicious software that infects a computer and restricts users' access to it until a ransom is paid to unlock it. Any failure by us to maintain or protect our information technology systems and data integrity, including from cyber-attacks, intrusions, or other breaches, could result in the unauthorized access to patient data and personally identifiable information, theft of intellectual property, or other misappropriation of assets, or otherwise compromise our confidential or proprietary information and disrupt our operations and could have a material adverse effect on our business, financial condition, and results of operations.
In the U.S., Federal and State privacy and security laws require certain of our operations to protect the confidentiality of personal information including patient medical records and other health information. In Europe, the Data Protection Directive requires us to manage individually identifiable information in the E.U. and, the GDPR may impose fines of up to four percent of our global revenue in the event of violations. Internationally, some countries have also passed laws that require individually identifiable data on their citizens to be maintained on local servers and that may restrict the transfer or processing of that data. We are also subject to the California Consumer Privacy Act (the “CCPA”), which went into effect in January 2020. In November 2020, California passed the California Privacy Rights Act (the “CPRA”), which builds on the CCPA and expands consumer privacy rights to more closely align with the GDPR. The CPRA went into effect on January 1, 2023, and applies to information collected on or after January 1, 2022. The CCPA and CPRA, among other things, create new data privacy obligations for covered companies and provides new privacy rights to California residents, including the right to opt out of certain disclosures of their information. The CCPA also created a private right of action with statutory damages for certain data breaches, thereby potentially increasing risks associated with a data breach. It remains unclear what, if any, additional modifications will be made to the CPRA by the California legislature or how it will be interpreted. We believe that we meet the expectations of applicable regulations and that the ongoing costs of compliance with such rules are not material to our business but could become material due to new regulations. There is no guarantee that we will be able to comply with these regulations, or otherwise avoid the negative reputational and other effects that might ensue from a significant data breach or failure to comply with applicable data privacy regulations, each of which could have significant adverse effects on our business, financial condition, or results of operations.
In recent years, companies around the world have seen a surge in wire transfer “phishing” attacks that attempt to trick employees into wiring money from company bank accounts to criminals’ bank accounts. In some cases, companies have lost millions of dollars to such relatively simple attacks, and these funds often are not recovered. While we take efforts to train employees to be cognizant of these types of attacks and take appropriate precautions, the level of technological sophistication used by attackers has increased in recent years, and a successful attack against us could lead to the loss of significant funds.
Although we possess insurance against the risk of cyber-attacks, there can be no assurance that the liability related to any such events will not exceed or insurance coverage limits or that such insurance will continue to be available on reasonable, commercially acceptable terms, or at all. If the costs of maintaining adequate insurance coverage should increase significantly in the future, our operating results could be materially adversely impacted.
The physical effects of climate change or legal, regulatory or market measures intended to address climate change could adversely affect our operations and operating results.
Shifts in weather patterns caused by climate change are expected over time to increase the frequency, severity, or duration of certain adverse weather conditions and natural disasters, such as hurricanes, tornadoes, earthquakes, wildfires, droughts, extreme temperatures or flooding, each of which could cause more significant business and supply chain interruptions, damage to our products and facilities as well as the infrastructure of hospitals, medical care facilities and other customers, reduced workforce availability, and increased costs of raw materials and components. While we do not expect climate change to materially affect the demand for our products, or the amount of persons with medical conditions we treat, climate change could also contribute to collateral effects such as increased transmission of viruses or airborne illnesses, which could contribute to unpredictable events, such as putting stress on hospital and other medical facilities and/or supply chains, and thus disrupting the elective surgery market in which we do business. In addition, increased public concern over climate change could result in new legal or regulatory requirements designed to mitigate the effects of climate change, which could include the adoption of more stringent environmental laws and regulations or stricter enforcement of existing laws and regulations. Such developments could result in increased compliance costs and adverse impacts on raw material sourcing, manufacturing operations and the distribution of our products, which could adversely affect our operations and operating results.
45
If any of our manufacturing, development or research facilities are damaged and/or our manufacturing processes are interrupted, we could experience supply disruptions, lost revenues and our business could be seriously harmed.
Damage to our manufacturing, development or research facilities or disruption to our business operations for any reason, including due to natural disaster (such as earthquake, wildfires and other fires or extreme weather), power loss, communications failure, unauthorized entry or other events, such as a flu or other health epidemic (such as the result of the COVID-19 pandemic), could cause us to discontinue development and/or manufacturing of some or all of our products for an undetermined period of time. The property damage and business interruption insurance coverage on these facilities that we maintain might not cover all losses under such circumstances, and we may not be able to renew or obtain such insurance in the future on acceptable terms with adequate coverage or at reasonable costs. If our facilities were damaged, they could be difficult to replace and could require substantial lead time to repair or replace. In particular, we manufacture certain of our biologics products in one facility in Irvine, California and any damage to that facility could adversely affect our ability to timely satisfy demand for those products. Out of an abundance of caution, in October 2020, we relocated part of our Biologics finished goods inventory from our Irvine facility to our Carlsbad office due to the threat of the Silverado Fire that was causing evacuations throughout Orange County, California. Disruptions to our business operations may result from damage to the facilities of, or disruption to the business operations of, our suppliers. For example, if we are unable to obtain disposables or other materials required to maintain “clean room” sterility in our Irvine facility, we may be unable to continue to manufacture products at that facility, which products accounts for a significant amount of our total revenue. Any significant disruption to our manufacturing operations and to our ability to meet market demand likely would have an adverse impact on our sales and revenues as key stakeholders, including our independent sales agents and stocking distributors and physician customers, transition to what they perceive as more reliable sources of products.
We depend on third-party manufacturers for many of our products.
We contract with third-party manufacturers to produce many of our products like many other companies in the medical device industry. If we or any such manufacturer fail to meet production and delivery schedules, it can have an adverse impact on our ability to sell such products. Further, whether we directly manufacture a product or utilize a third-party manufacturer, shortages and spoilage of materials, labor stoppages, product recalls, manufacturing defects, and other similar events can delay production and inhibit our ability to bring a new product to market in timely fashion. For example, the supply of the Trinity ELITE and Trinity Evolution allografts are derived from human cadaveric donors, and our ability to market the tissues depends on MTF continuing to have access to donated human cadaveric tissue and their continued maintenance of high standards in their processing methodology.
We depend on a limited number of third-party suppliers for processing activities, components and raw materials and losing any of these suppliers, or their inability to provide us with an adequate supply of materials that meet our quality and other requirements, could harm our business.
Outside suppliers, some of whom are sole-source suppliers, provide us with products and raw materials and components used in manufacturing our biologics and spinal implant products. We strive to maintain sufficient inventory of products, raw materials and components so that our production will not be significantly disrupted if a particular product, raw material or component is not available to us for a period of time, including as a result of a supplier's loss of its ISO or other certification or as a result of any of the disruptions described below under the risk factor titled “If any of our manufacturing, development or research facilities are damaged and/or our manufacturing processes are interrupted, we could experience supply disruptions, lost revenues and our business could be seriously harmed.” For example, a certain number of our products require titanium, which is sourced from third party suppliers. While the titanium required for such products is not directly sourced from Russia, the current geopolitical events involving Russia and Ukraine is negatively impacting the wider titanium supply chain and such geopolitical events and factors relating thereto or resulting therefrom, including the imposition of sanctions, may negatively impact the ability of our local supply sources to timely supply titanium to us. In addition, some of our suppliers may choose to discontinue making their products available in the EU rather than follow MDR, which would require us to identify alternate supply sources for those products. Any such disruption in our production could harm our reputation, business, financial condition and results of operations.
Although we believe there are alternative supply sources, replacing our suppliers may be impractical or difficult in many instances. For example, we could have difficulty obtaining similar services or products from other suppliers that are acceptable to the FDA or other foreign regulatory authorities and who are able to provide the appropriate supply volumes at an acceptable cost. In addition, if we are required to transition to new suppliers for certain services or components of our products, the use of services, components, or materials furnished by these alternative suppliers could require us to alter our operations, and if we are required to change the manufacturer of a critical component of our products, we will have to verify that the new manufacturer maintains facilities, procedures and operations that comply with our quality and applicable regulatory requirements, which could further impede our ability to manufacture our products in a timely manner. Transitioning to a new supplier could be time-consuming and expensive,
46
may result in interruptions in our operations and product delivery, could affect the performance specifications of our products, or could require that we modify the design of those systems.
If we are unable to obtain sufficient quantities of spinal implant products, raw materials or components that meet our quality and other requirements on a timely basis for any reason, we may not produce sufficient quantities of our products to meet market demand until a new or alternative supply source is identified and qualified and, as a result, we could lose customers, our reputation could be harmed and our business could suffer. Furthermore, an uncorrected defect or supplier’s variation in a component or raw material that is incompatible with our manufacturing, or unknown to us, could harm our ability to manufacture products.
Further, under the FDASIA, which includes the Medical Device User Fee Amendments of 2012, as well as other medical device provisions, all U.S. and foreign manufacturers must have a FDA Establishment Registration and complete Medical Device listings for sales in the U.S. While we believe that our facilities materially comply with these requirements, we also source products from foreign contract manufacturers. It is possible that some of our foreign contract manufacturers will not comply with applicable requirements and choose not to register with the FDA. In such an event, we will need to determine if there are alternative foreign contract manufacturers who comply with the applicable requirements. If such a foreign contract manufacturer is a sole supplier of one of our products, there is a risk that we may not be able to source another supplier.
Furthermore, we rely on a small number of tissue banks accredited by the American Association of Tissue Banks for the supply of human tissue, a crucial component of our biologics products that serve as bone graft substitutes. Any failure to obtain tissue from these sources or to have the tissue processed by these sources for us in a timely manner will interfere with our ability to meet demand for our biologics products effectively. The processing of human tissue into biologics products is labor intensive and maintaining a steady supply stream is challenging. In addition, due to seasonal changes in mortality rates, some scarce tissues used for our biologics products are at times in particularly short supply. If governments require additional donor testing due to COVID-19, this could also strain the supply of tissue. We cannot be certain that our supply of human tissue from our suppliers will be available at current levels or will meet our needs or that we will be able to successfully negotiate commercially reasonable terms with other accredited tissue banks.
If we are unable to maintain and expand our network of independent sales representatives and distributors, we may not maintain or grow our revenue.
We sell our products in many countries through independent sales representatives and distributors. Frequently, our independent sales representatives and our distributors have the exclusive right to sell our products in their respective territories. If any of our independent sales representatives or distributors fail to adequately promote, market and sell our products, our sales could significantly decrease. The terms of our agreements with our independent sales representatives and distributors vary in length, generally from one to ten years. Under the terms of our standard distribution agreements, each party has the right to terminate in the event of a material breach by the other party and we generally have the right to terminate if the distributor does not meet agreed sales targets or fails to make payments on time. Any termination of our existing relationships with independent sales representatives or distributors could have an adverse effect on our business unless and until commercially acceptable alternative distribution arrangements are put in place. In addition, we operate in areas of the world that have been or may be disproportionately affected by recessions or disasters and we bear risk that existing or future accounts receivable may be uncollected if these distributors or hospitals experience disruptions to their business that cause them to discontinue paying ongoing accounts payable or become insolvent.
Further, we face significant challenges and risks in managing our geographically dispersed distribution network and retaining the independent sales representatives and distributors who make up that network, and as we launch new products and increase our marketing efforts with respect to existing products, we plan to expand the reach of our marketing and sales efforts and may need to hire new independent sales representatives and distributors. Independent sales representatives and distributors require significant technical expertise in various areas such as spinal care practices, spine injuries and disease, and spinal health and they require training and time to achieve full productivity. We may not attract or retain qualified independent sales representatives and distributors or enter into agreements with them on favorable or commercially reasonable terms, if at all. This could be due to a number of factors, including, but not limited to, perceived deficiencies, or gaps, in our existing product portfolio, intense competition for services of independent sales representatives and distributors, or because of the disruption associated with restrictive covenants to which representatives or distributors may be subject and potential litigation and expense associated therewith. We may also experience unforeseen disengagement from independent sales representatives and distributors who have worked with us for many years. Even if we enter into agreements with additional qualified independent sales representatives or distributors, it often takes 6 to 12 months for new sales representatives or distributors to reach full operational effectiveness and they may not generate revenue as quickly as we expect them to, commit the necessary resources to effectively market and sell our products, or ultimately succeed in selling our products. Our success will depend largely on our ability to continue to hire, train, retain
47
and motivate qualified independent sales representatives and distributors. If we cannot expand our sales and marketing capabilities domestically and internationally, if we fail to train new independent sales representatives and distributors adequately, or if we experience high turnover in our sales network, we may not commercialize our products adequately, or at all, which would adversely affect our business, results of operations and financial condition.
Moreover, because our independent sales representatives and distributors are not our employees, we have limited control over their activities and, generally, we do not enter into exclusive relationships with them. If one or more of them were to be retained by a competitor, whether or an exclusive or non-exclusive basis, they may divert business from us to our competitor, which could materially and adversely affect our sales.
We depend on our senior management team.
Our success depends upon the skill, experience, and performance of members of our senior management team, who have been critical to the management of our operations and the implementation of our business strategy. We do not have key man insurance on our senior management team, and the loss of one or more key executive officers could have a material adverse effect on our operations. Further, any turnover in our senior management team could adversely affect our operating results and cash flows.
In order to compete, we must attract, retain, and motivate key employees, and our failure to do so could have an adverse effect on our results of operations.
In order to compete, we must attract, retain, and motivate executives and other key employees, including those in managerial, technical, sales, marketing, research, development, finance, information and technology, and other support positions representing diverse backgrounds, experiences, and skill sets. Hiring and retaining qualified executives, engineers, technical staff, and sales representatives is critical to our business, and competition for experienced employees in the medical device industry can be intense. Maintaining our brand and reputation, as well as a diverse and inclusive work environment that enables all our employees to thrive, are important to our ability to recruit and retain employees. If we are less successful in our recruiting efforts, or if we cannot retain highly skilled workers and key leaders, our ability to develop and deliver successful products and services may be adversely affected.
Moreover, replacing key employees may be a difficult, costly, and protracted process, and we may not have other personnel with the capacity to assume all of the responsibilities of a departing employee. Competition for qualified personnel, particularly for key positions, is intense among companies in our industry, and many of the organizations against which we compete for qualified personnel have greater financial and other resources and different risk profiles than our company, which may make them more attractive employers. All of our employees, including our management personnel, may terminate their employment with us at any time without notice. If we cannot attract and retain highly qualified personnel, as needed, we may not achieve our financial and other goals.
To attract, retain, and motivate qualified executives and key employees, we utilize stock-based incentive awards, such as employee stock options, and restricted stock units. Certain awards vest based upon the passage of time while others vest upon the achievement of certain performance-based or market-based conditions. If the value of such stock awards does not appreciate, as measured by the performance of the price of our common stock, and ceases to be viewed as a valuable benefit, our ability to attract, retain, and motivate our employees could be adversely impacted, which could negatively affect our results of operations and/or require us to increase the amount we expend on cash and other forms of compensation.
In addition, future internal growth could impose significant added responsibilities on our management, and we will need to identify, recruit, maintain, motivate, and integrate additional employees to manage growth effectively. If we do not effectively manage such growth, our expenses may increase more than expected, we may not achieve our goals, and our ability to generate and/or grow revenue could be diminished.
Our business is subject to economic, political, regulatory, and other risks associated with international sales and operations.
Because we sell our products in many different countries, our business is subject to risks associated with conducting business internationally. We anticipate that net sales from international operations will continue to represent a substantial portion of our total net sales. In addition, certain of our manufacturing facilities and suppliers are located outside the U.S. Accordingly, our future results could be harmed by a variety of factors, including:
•changes in a specific country’s or region’s political, social, or economic conditions;
•difficulties in staffing and managing widespread operations;
48
•having to comply with export control laws, including, but not limited to, the Export Administration Regulations and trade sanctions against embargoed countries, which are administered by the Office of Foreign Assets Control within the Department of the Treasury, as well as the laws and regulations administered by the Department of Commerce;
•complex data privacy requirements, including, but not limited to, the GDPR;
•differing regulatory requirements for obtaining clearances or approvals to market our products, and unexpected changes in regulatory requirements;
•changes in, or uncertainties relating to, foreign rules and regulations that may impact our ability to sell our products, perform services or repatriate profits to the U.S.;
•tariffs, trade barriers and export regulations that adversely impact, and other regulatory and contractual limitations on, our ability to sell our products in certain foreign markets, the scope and consequences of which are subject to changing agendas of political, business and environmental groups;
•consequences from changes in tax or customs laws;
•fluctuations in foreign currency exchange rates;
•limitations on or increase of withholding and other taxes on remittances and other payments by foreign subsidiaries or joint ventures;
•differing multiple payer reimbursement regimes, government payers or patient self-pay systems;
•differing labor laws and standards;
•an inability, or reduced ability, to protect our intellectual property, including any effect of compulsory licensing imposed by government action;
•availability of government subsidies or other incentives that benefit competitors in their local markets that are not available to us; and
•having to comply with various U.S. and international laws, including the FCPA and anti-money laundering laws, and violation by our independent sales representatives or distributors of such laws.
Risks Related to our Intellectual Property
We depend on our ability to protect our intellectual property and proprietary rights, but we may not be able to maintain the confidentiality of these assets or assure their protection.
Our success depends, in large part, on our ability to protect our current and future technologies and products and to defend our intellectual property rights. If we fail to protect our intellectual property adequately, competitors may manufacture and market products that are similar to, or that compete directly with, our products. Numerous patents covering our technologies have been issued to us and we have filed, and expect to continue to file, patent applications seeking to protect newly developed technologies and products in various countries, including the U.S. Some patent applications in the U.S. are maintained in secrecy until the patent is issued. Because the publication of discoveries tends to follow their actual discovery by several months, we may not be the first to invent or file patent applications on any of our discoveries. Further, there is a substantial backlog of patent applications at the U.S. Patent and Trademark Office, and the approval or rejection of patent applications may take several years. Patents may not be issued with respect to any of our patent applications and existing or future patents issued to or licensed by us and may not provide adequate protection or competitive advantages for our products. Patents that are issued may be challenged, invalidated, or circumvented by our competitors. Furthermore, our patent rights may not prevent our competitors from developing, using, or commercializing products that are similar or functionally equivalent to our products. Moreover, if patents are not issued with respect to our products arising from research, we may not be able to maintain the confidentiality of information relating to these products. In addition, if a patent relating to any of our products lapses or is invalidated, we may experience greater competition arising from new market entrants.
We also rely on trade secrets, unpatented proprietary expertise, and continuing technological innovation that we protect, in part, by entering into confidentiality agreements with assignors, licensees, suppliers, employees, and consultants. These agreements may be breached and there may not be adequate remedies in the event of a breach. Disputes may arise concerning the ownership of intellectual property or the applicability or enforceability of confidentiality agreements. Moreover, our trade secrets and proprietary technology may otherwise become known or be independently developed by our competitors.
49
In addition to contractual measures, we try to protect the confidential nature of our proprietary information using physical and technological security measures. Such measures may not, for example, in the case of misappropriation of a trade secret by an employee or third party with authorized access, adequately protect our proprietary information. Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Unauthorized parties may also attempt to copy or reverse engineer certain aspects of our products that we consider proprietary. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive, and time-consuming, and the outcome is unpredictable.
We may face claims by third parties that our agreements with employees, consultants or advisors obligating them to assign intellectual property to us are ineffective or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such intellectual property. Litigation may be necessary to resolve an ownership dispute, and if we are unsuccessful, we may be precluded from using certain intellectual property or may lose our exclusive rights in that intellectual property. Either outcome could harm our business and competitive position.
Furthermore, the laws of some foreign countries may not protect our intellectual property rights to the same extent as the laws of the U.S., if at all. Since certain of our issued patents and pending patent applications are for the U.S. only, we lack a corresponding scope of patent protection in other countries. Thus, we may not be able to stop a competitor from marketing products in other countries that are similar to some of our products.
If we are unable to obtain, protect and enforce patents on our technology and to protect our trade secrets, such inability could have a material and adverse effect on our business, results of operations, and financial condition.
Third parties may claim that we infringe on their proprietary rights and may prevent us from manufacturing and selling certain of our products.
Our success will depend in part on our ability, both in the U.S. and in foreign countries, to operate without infringing upon the patents and proprietary rights of others, and to obtain appropriate licenses to patents or proprietary rights held by third parties if infringement would otherwise occur.
There has been substantial litigation in the medical device industry with respect to the manufacture, use, and sale of new products. These lawsuits relate to the validity and infringement of patents or proprietary rights of third parties. We may be required to defend against allegations relating to the infringement of patent or proprietary rights of third parties. Any such litigation could, among other things:
•Require us to incur substantial expense, even if we are successful in the litigation;
•Require us to divert significant time and effort of our technical and management personnel;
•Result in the loss of our rights to develop or make certain products; and
•Require us to pay substantial monetary damages or royalties in order to license proprietary rights from third parties or to satisfy judgments or to settle actual or threatened litigation.
Although patent and intellectual property disputes within the medical devices industry have often been settled through assignments, licensing, or similar arrangements, costs associated with these arrangements may be substantial and could include the long-term payment of royalties. Accordingly, an adverse determination in a judicial or administrative proceeding, or a failure to obtain necessary assignments or licenses, could result in us having to pay substantial damages (which may be increased up to three times of awarded damages) and/or substantial royalties, and could prevent us from manufacturing or selling some products or increase our costs to market these products unless we obtain a license or are able to redesign our products to avoid infringement. Any such license may not be available on reasonable terms, if at all, and there can be no assurance that we would be able to redesign our products in a way that would not infringe the intellectual property rights of others. If we fail to obtain any required licenses or make any necessary changes to our products or technologies, we may have to withdraw existing products from the market or may be unable to commercialize one or more of our products, all of which could have a material adverse effect on our business, results of operations and financial condition.
In addition, we generally indemnify our customers and sales representatives with respect to infringement by our products of the proprietary rights of third parties. Third parties may assert infringement claims against our customers or sales representatives. These claims may require us to initiate or defend protracted and costly litigation on behalf of our customers or sales representatives, regardless of the merits of these claims. If any of these claims succeed, we may be forced to indemnify, or pay damages on behalf of,
50
our customers or sales representatives or may have to obtain licenses for the products they use. If we cannot obtain all necessary licenses on commercially reasonable terms, our customers may be forced to stop using our products.
If we seek to protect or enforce our intellectual property rights through litigation or other proceedings, it could require us to spend significant time and money, the results of which are uncertain.
To protect or enforce our intellectual property rights, we may have to initiate or defend litigation against or by third parties, such as infringement suits, opposition proceedings, or seeking a court declaration that we do not infringe the proprietary rights of others or that their rights are invalid or unenforceable. We may not have sufficient resources to enforce our intellectual property rights or to defend our intellectual property rights against a challenge. Even if we prevail, the cost of litigation, including the diversion of management and other resources, could affect our profitability and could place a significant strain on our financial resources.
Our ability to enforce our intellectual property rights depends on our ability to detect infringement. It may be difficult to detect infringers who do not advertise the components used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product. The medical device industry is characterized by the existence of a large number of patents and frequent litigation based on allegations of patent infringement. It is not unusual for parties to exchange letters surrounding allegations of intellectual property infringement and licensing arrangements. In addition, the patent positions of medical device companies, including our patent position, may involve complex legal and factual questions, and, therefore, the scope, validity and enforceability of any patent claims we have or may obtain cannot be predicted with certainty.
We may be subject to claims that we, our employees, or our independent sales agents or stocking distributors have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition or non-solicitation agreements with our competitors.
Many of our employees were employed at other medical device companies, including our competitors or potential competitors, in some cases immediately prior to joining us. In addition, many of our independent sales representatives and distributors sell, or in the past have sold, products of our competitors. We may be subject to claims that we, our employees or our independent sales representatives or distributors intentionally, inadvertently or otherwise used or disclosed trade secrets or other proprietary information of former employers or competitors. In addition, we have been and may in the future be subject to claims that we caused an employee, or encouraged/assisted an independent sales agent, to breach the terms of his or her non-competition or non-solicitation agreement. Litigation may be necessary to defend against these claims. Litigation is expensive and time-consuming, and could divert management attention and resources away from our business. Even if we prevail, the cost of litigation could affect our profitability. If we do not prevail, in addition to any damages we might have to pay, we may lose valuable intellectual property rights or employees, independent sales representatives or distributors. There can be no assurance that this type of litigation or the threat thereof will not adversely affect our ability to engage and retain key employees, sales representatives or distributors.
Risks Related to Litigation and Product Liability Matters
We may be subject to product and other liability claims that may not be covered by insurance and could require us to pay substantial sums.
We are subject to an inherent risk of, and adverse publicity associated with, product liability and other liability claims, whether or not such claims are valid. Spine surgery involves significant risk of serious complications, including bleeding, nerve injury, paralysis and even death. In addition, if neurosurgeons and orthopedic spine surgeons are not sufficiently trained in the use of our products, they may misuse or ineffectively use our products, which may result in unsatisfactory patient outcomes or patient injury. We could become the subject of product liability lawsuits alleging that component failures, malfunctions, manufacturing flaws, design defects, or inadequate disclosure of product-related risks or product-related information resulted in an unsafe condition or injury to patients. In addition, the development of allograft implants and technologies for human tissue repair and treatment may entail particular risk of transmitting diseases to human recipients, and any such transmission could result in the assertion of product liability claims against us.
Product liability claims are expensive to defend, divert our management’s attention and, if we are not successful in defending the claim, can result in substantial monetary awards against us or costly settlements. Further, successful product liability claims made against one or more of our competitors could cause claims to be made against us or expose us to a perception that we are vulnerable to similar claims. Any product liability claim brought against us, with or without merit and regardless of the outcome or whether it is fully pursued, may result in: decreased demand for our products; injury to our reputation; significant litigation costs; product recalls; loss of revenue; the inability to commercialize new products or product candidates; and adverse publicity regarding our products. Any of these may have a material and adverse effect on our reputation with existing and potential customers and on
51
our business, financial condition, and results of operations. In addition, a recall of some of our products, whether or not the result of a product liability claim, could result in significant costs and loss of customers.
We maintain product liability insurance coverage in amounts and scope that we believe are reasonable and adequate. There can be no assurance, however, that product liability or other claims will not exceed our insurance coverage limits or that such insurance will continue to be available on reasonable, commercially acceptable terms, or at all. A successful product liability claim that exceeds our insurance coverage limits could require us to pay substantial sums and could have a material adverse effect on our financial condition. In addition, a recall of some of our products, whether or not the result of a product liability claim, could result in significant costs and loss of customers.
Our insurance policies are expensive and protect us only from some risks, which will leave us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk to which our business is or may be exposed. Some of the policies we maintain include product liability insurance, directors’ and officers’ liability insurance, property insurance, and workers’ compensation insurance. We do not know, however, if we will be able to maintain insurance coverage at a reasonable cost or in sufficient amounts or scope to protect us against losses. If the costs of maintaining adequate insurance coverage should increase significantly in the future, our operating results could be materially adversely impacted. Even if we have insurance, a claim could exceed the amount of our insurance coverage or it may be excluded from coverage under the terms of the policy. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our cash position and results of operations.
Risks Related to Potential Acquisitions, Investments, and Divestitures
Our efforts to identify, pursue, and implement new business opportunities (including acquisitions) may be unsuccessful and may have an adverse effect on our business.
Our growth depends, in large part, on our ability to identify, pursue, and implement new business opportunities that expand our product offerings, capabilities, and geographic presence, and we compete with other medical device companies for these opportunities. Our efforts to identify such opportunities focus primarily on potential acquisitions of new businesses, products or technologies, licensing arrangements, commercialization arrangements, and other transactions with third parties. We may not be able to identify business opportunities that meet our strategic criteria or that are acceptable to us or our stockholders. Even if we are able to identify acceptable business opportunities, we may not be able to pursue or implement such business opportunities (or, in the case of acquisitions or other transactions, complete such acquisitions or other transactions) in a timely manner or on a cost-effective basis (or at all), and we may not realize the expected benefits of such business opportunities. If we are not able to identify, pursue, and implement new business opportunities, it will adversely affect our ability to grow our business.
In addition, pursuing and implementing new business opportunities (particularly acquisitions) may involve significant costs and entail risks, uncertainties, and disruptions to our business, especially where we have limited experience as a company developing or marketing a particular product or technology or operating in a particular geographic region. We may be unable to integrate a new business, product, or technology effectively, or we may incur significant charges related to an acquisition or other business opportunity (for example, amortization of acquired assets or asset impairment charges), which may adversely affect our business, financial condition, and results of operations. Newly acquired technology or products may require additional development efforts prior to commercial sale, including clinical testing and approval by the FDA and applicable foreign regulatory authorities; such additional development efforts may involve significant expense and ultimately be unsuccessful. Any cross-border acquisitions or transactions may involve unique risks in addition to those mentioned above, including those related to integration of operations across different cultures and languages, currency risks, and the particular economic, political, and regulatory risks associated with specific countries. To the extent we issue additional equity in connection with acquisitions, this may dilute our existing stockholders.
Furthermore, as a result of acquisitions of other healthcare businesses, we may be subject to the risk of unanticipated business uncertainties, regulatory and other compliance matters or legal liabilities relating to those acquired businesses for which the sellers of the acquired businesses may not indemnify us, for which we may not be able to obtain insurance (or adequate insurance) or for which the indemnification may not be sufficient to cover the ultimate liabilities.
We have provided over $10.0 million in investments and loans to a privately-held company in Switzerland and may not be able to recoup our investment.
In October 2020, we entered into agreements with Neo Medical SA, a privately-held Swiss-based medical technology company developing a new generation of products for spinal surgery (“Neo Medical”). Our collaboration with Neo Medical focuses on co-developing with them a cervical platform and deploying single-use, sterile-packed procedure solutions designed to increase
52
operating room efficiencies, reduce procedural times and costs, improve patient outcomes through novel device designs and techniques, and reduce infection rates. These instruments are designed for surgical settings including acute care hospitals, outpatient hospitals, and also ambulatory surgery centers. Under our agreements with Neo Medical, we will also exclusively distribute Neo Medical’s thoracolumbar procedure solutions to certain U.S. accounts.
In connection with these arrangements, we purchased $5.0 million of Neo Medical’s preferred stock, and loaned CHF 4.6 million ($5.0 million as of the issuance date) to Neo Medical pursuant to a convertible loan agreement. The loan accrues interest at an annual rate of 8% and is convertible by either party into additional shares of Neo Medical’s preferred stock. If not otherwise converted to preferred stock in the interim, the loan and all accrued interest become due and payable in October 2024. In October 2021, the Company entered into an additional Convertible Loan Agreement (the “Additional Convertible Loan”), pursuant to which the Company loaned Neo Medical an additional CHF 0.6 million ($0.7 million as of the issuance date). In January 2022, the Company elected to convert the Additional Convertible Loan into shares of Neo Medical’s preferred stock.
Neo Medical is using the proceeds of our preferred stock purchase and loans to fund its ongoing operations. However, no assurance can be made that Neo Medical’s business ultimately will be successful. As such, we could ultimately be unable to recoup any value for the preferred stock that we purchased and/or unable to recoup the amount of our loan.
We may incur significant costs or retain liabilities associated with disposition activity.
We may from time to time sell, license, assign, or otherwise dispose of or divest assets, the stock of subsidiaries, or individual products, product lines, or technologies, which we determine are no longer desirable for us to own, some of which may be material. Any such activity could result in us incurring costs and expenses from these efforts, some of which could be significant. This may also result in us retaining liabilities related to the assets or properties disposed of even though, for instance, the income-generating assets have been disposed. These costs and expenses may be incurred at any time and may have a material impact on our results of operations.
Risks Related to Our Financial Results and Need for Financing
Our quarterly operating results may fluctuate.
Our quarterly operating results have fluctuated significantly in the past. Our future quarterly operating results may fluctuate significantly and we may experience losses depending on a number of factors, many of which are outside our control. Such factors include:
•economic conditions worldwide, including arising from or relating to the effects of the COVID-19 pandemic, which could affect the ability of hospitals and other customers to purchase our products and could result in a reduction in elective and non-reimbursed operative procedures;
•market acceptance of our existing products, as well as products in development, and the demand for, and pricing of, our products and the products of our competitors;
•costs, benefits and timing of new product introductions;
•the timing of or failure to obtain regulatory clearances or approvals for new products;
•lost sales and other expenses resulting from stoppages in our or third parties’ production, including as a result of product recalls or field corrective actions;
•the availability and cost of components and materials, including raw materials such as human tissue;
•accurate predictions of product demand and production capabilities sufficient to meet that demand;
•our ability to realize expected yield improvements and scrap reduction initiatives that we have undertaken at our Irvine facility;
•higher than anticipated independent sales representatives and distributors commissions;
•our ability to purchase or manufacture and ship our products efficiently and in sufficient quantities to meet sales demands;
•the timing of our research and development expenditures;
53
•expenditures for major initiatives;
•the timing and level of reimbursement, changes in reimbursement or denials in coverage for our products by third-party payors, such as Medicare, Medicaid, private and public health insurers and foreign governmental health systems;
•the ability of our independent sales representatives and distributors to achieve expected sales targets and for new agents and stocking distributors to become familiar with our products in a timely manner;
•peer-reviewed publications discussing the clinical effectiveness of our products;
•inspections of our manufacturing facilities for compliance with the FDA's Quality System Regulations (Good Manufacturing Practices), which could result in Form 483 observations, warning letters, injunctions or other adverse findings from the FDA or equivalent foreign regulatory bodies, and corrective actions, procedural changes and other actions, including product recalls, that we determine are necessary or appropriate to address the results of those inspections, any of which may affect production and our ability to supply our customers with our products;
•the costs to comply with new regulations from the FDA or equivalent foreign regulatory bodies, such as the requirements to establish a unique device identification system to adequately identify medical devices through their distribution and use;
•the increased regulatory scrutiny of certain of our products, including products we manufacture for others, which could result in their being removed from the market;
•fluctuations in foreign currency exchange rates; and
•the impact of acquisitions, including the impact of goodwill and intangible asset impairment charges, if future operating results of the acquired businesses are significantly less than the results anticipated at the time of the acquisitions.
In addition, we may experience meaningful variability in our sales and gross profit among quarters, as well as within each quarter, as a result of several factors, including but not limited to (and in addition to those listed above):
•the number of products sold in the quarter;
•the unpredictability of sales of full sets of spinal implants and instruments to our international stocking distributors; and
•the number of selling days in the quarter.
Our goodwill, intangible assets and fixed assets are subject to potential impairment; we have recorded significant goodwill impairment charges and may be required to record additional charges to future earnings if our remaining goodwill or intangible assets become impaired.
A significant portion of our assets consists of goodwill, intangible assets and fixed assets. The carrying value of these assets may be reduced if we determine that those assets are impaired, including intangible assets from recent acquisitions.
Most of our intangible and fixed assets have finite useful lives and are amortized or depreciated over their useful lives on a straight-line basis. The underlying assumptions regarding the estimated useful lives of these intangible assets are analyzed on at least an annual basis and more often if an event or circumstance occurs making it likely that the carrying value of the assets may not be recoverable. Any such changes are adjusted through accelerated amortization, if necessary. Whenever events or changes in circumstances indicate that the carrying value of the assets may not be recoverable, we test intangible assets for impairment based on estimates of future cash flows. Factors that may be considered a change in circumstances indicating that the carrying value of our intangible assets and/or goodwill may not be recoverable include a decline in stock price and market capitalization, slower growth rates in our industry, the introduction of newer technology or competing products that may cannibalize future sales, or other materially adverse events that have implications on the profitability of our business. When testing for impairment of finite-lived intangible assets held for use, we group assets at the lowest level for which cash flows are separately identifiable. If an intangible asset is considered to be impaired, the amount of the impairment will equal the excess of the carrying value over the fair value of the asset.
Goodwill is required to be tested for impairment at least annually. We review our two reporting units for potential goodwill impairment in the fourth fiscal quarter of each year as part of our annual goodwill impairment testing, and more often if an event or circumstance occurs making it likely that impairment exists. During the fourth quarter of 2021, we recorded a full impairment of the Global Orthopedics goodwill. This resulted in an impairment charge of $11.8 million, which is reflected within acquisition-related amortization and remeasurement on the Consolidated Statement of Operations. If actual results differ from the assumptions and
54
estimates used in the goodwill and intangible asset calculations, we could incur future impairment or amortization charges, which could negatively impact our financial condition and results of operations.
We face risks related to foreign currency exchange rates.
Because some of our revenue, operating expenses, assets, and liabilities are denominated in foreign currencies, we are subject to foreign exchange risks that could adversely affect our operations and reported results. To the extent that we incur expenses or recognize net sales in currencies other than the U.S. Dollar, any change in the values of those foreign currencies relative to the U.S. Dollar could cause our profits to decrease or our products to be less competitive against those of our competitors. To the extent that our current assets denominated in foreign currency are greater or less than our current liabilities denominated in foreign currencies, we have potential foreign exchange exposure. The fluctuations of foreign exchange rates during 2022 had an unfavorable impact of $10.5 million on net sales outside of the U.S. Although we seek to manage our foreign currency exposure by matching non-dollar revenues and expenses, exchange rate fluctuations could have a material adverse effect on our results of operations in the future. To minimize such exposures, we may enter into currency hedges from time to time.
In addition, for those foreign customers who purchase our products in U.S. Dollars, currency exchange rate fluctuations between the U.S. Dollar and the currencies in which those customers do business may have a negative effect on the demand for our products in foreign countries where the U.S. Dollar has increased in value compared to the local currency. Converting our earnings from international operations to U.S. Dollars for use in the U.S. can also raise challenges, including problems moving funds out of the countries in which the funds were earned and difficulties in collecting accounts receivable in foreign countries where the usual accounts receivable payment cycle is longer.
Our global operations may expose us to tax risks.
We are subject to taxes in the U.S. and numerous foreign jurisdictions. Significant judgment and interpretation of tax laws are required to estimate our tax liabilities. Tax laws and rates in various jurisdictions may be subject to significant change as a result of political and economic conditions. Our effective income tax rate could be adversely affected by changes in those tax laws, changes in the mix of earnings among tax jurisdictions, changes in the valuation of our deferred tax assets and liabilities, vesting of equity awards at a price below the original valuation, historical entity classification elections, and the resolution of matters arising from tax audits.
Beginning in 2022, the Tax Cuts and Jobs Act of 2017 eliminated the option to deduct research and development expenditures immediately in the year incurred and requires taxpayers to amortize such expenditures over five years, or 15 years for such expenditures incurred outside of the U.S. This requirement may have a significant impact on our cash tax liability and our effective tax rate as we perform research and development in the U.S., Italy, and Canada.
Certain of our subsidiaries sell products directly to other Orthofix subsidiaries or provide marketing and support services to other Orthofix subsidiaries. These intercompany sales and support services involve subsidiaries operating in jurisdictions with differing tax rates and we must determine the appropriate allocation of income to each jurisdiction based on current interpretations of complex income tax regulations. Tax authorities in these jurisdictions may challenge our treatment of such intercompany transactions. If we are unsuccessful in defending our treatment of intercompany transactions, we may be subject to additional tax liability, interest, or penalty, which could adversely affect our profitability.
We maintain a $300.0 million secured revolving credit facility secured by a pledge of substantially all of our property.
In October 2019, we and certain of our wholly-owned subsidiaries (collectively, the “Borrowers”) entered into a Second Amended and Restated Credit Agreement (the “Amended Credit Agreement”). The Amended Credit Agreement provides for a $300.0 million secured revolving credit facility maturing on October 25, 2024, and amends and restates the previous $125.0 million secured revolving credit facility. No amount is currently outstanding on the credit facility as of December 31, 2022, or as of the date hereof, but we may draw on this facility in the future.
Certain of our subsidiaries (collectively, the “Guarantors”) are required to guarantee the repayment of any obligations under the Amended Credit Agreement. The obligations with respect to the Amended Credit Agreement are secured by a pledge of substantially all of the personal property assets of the Borrowers and each of the Guarantors, including accounts receivables, deposit accounts, intellectual property, investment property, and inventory, equipment, and equity interests in their respective subsidiaries.
The Amended Credit Agreement contains customary affirmative and negative covenants, including limitations on our ability to incur additional debt, grant or permit additional liens, make investments and acquisitions, merge or consolidate with others, dispose of assets, pay dividends and distributions, pay subordinated indebtedness, and enter into affiliate transactions. In addition, the Amended Credit Agreement contains financial covenants requiring us to maintain, on a consolidated basis as of the last day of any
55
fiscal quarter, a total net leverage ratio of not more than 3.5 to 1.0 (which ratio can be permitted to increase to 4.0 to 1.0 for no more than 4 fiscal quarters following a material acquisition) and an interest coverage ratio of at least 3.0 to 1.0. The Amended Credit Agreement also includes events of default customary for facilities of this type and upon the occurrence of such events of default, subject to customary cure rights, all outstanding loans under the facility may be accelerated and/or the lenders’ commitments terminated.
We believe that we are in compliance with the covenants, and there were no events of default, at December 31, 2022 (and in prior periods). However, there can be no assurance that we will be able to meet such financial covenants in future fiscal quarters. The failure to do so could result in an event of default under such agreement, which could have a material adverse effect on our financial position in the event that we have significant amounts drawn under the facility at such time.
We must maintain high levels of inventory, which could consume a significant amount of our resources and reduce our cash flows.
Because we maintain substantial inventory levels to meet the needs of our customers, we are subject to the risk of inventory excess, obsolescence and shelf-life expiration. Many of our spinal implant products come in sets. Each set includes a significant number of components in various sizes so that the physician may select the appropriate spinal implant based on the patient’s needs. In a typical surgery, not all of the implants in the set are used, and therefore certain sizes of implants placed in the set or that we purchase for replenishment inventory may become obsolete before they can be used. In addition, to market our products effectively, we often must provide hospitals and independent sales agents with consigned sets that typically consist of spinal implants and instruments, including products to ensure redundancy and products of different sizes. Further, our biologics products have expiration dates, which range from one to five years, and these products may expire before they can be used. If a substantial portion of our inventory is deemed excess, becomes obsolete, or expires, it could have a material adverse effect on our earnings and cash flows due to the resulting costs associated with the inventory impairment charges and costs required to replace such inventory. Further, as we increasingly launch new products and product systems, we may cannibalize older products and product systems, which could exacerbate excess and obsolete charges.
We may need additional financing in the future to meet our capital needs or to make opportunistic acquisitions, and such financing may not be available on favorable terms, if at all.
We may need additional financing in the future to meet our capital needs or to make opportunistic acquisitions. The capital and credit markets may experience extreme volatility and disruption, which may lead to uncertainty and liquidity issues for both borrowers and investors, and we may be unable to obtain any desired additional financing on favorable terms, if at all. If adequate funds are not available to us on acceptable terms, we may be unable to successfully develop or enhance products, or respond to competitive pressures, any of which could negatively affect our business, results of operations and financial condition. If we raise capital by issuing debt or entering into credit facilities, we may be subject to limitations on our operations due to restrictive covenants.
General Risks
Our stock price has fluctuated and may continue to fluctuate, which may make future prices of our stock difficult to predict.
Investors should not rely on recent or historical trends to predict future stock prices, financial condition, results of operations, or cash flows. Our stock price, like that of other medical device companies, can be volatile and can be affected by, among other things: speculation, coverage, or sentiment in the media or the investment community; the announcement of new, planned or contemplated products, services, technological innovations, acquisitions, divestitures, or other significant transactions by us or our competitors; our quarterly financial results and comparisons to estimates by the investment community or financial outlook provided by us; the financial results and business strategies of our competitors; publication of research reports about us or our industry or changes in recommendations or withdrawal of research coverage by securities analysts; changes in laws or regulations affecting our business, including tax legislation; changes in accounting standards, policies, guidance, interpretations or principles; threatened or actual litigation or governmental investigations; and inflation; market volatility or downturns caused by outbreaks, epidemics, pandemics, geopolitical tensions or conflicts, or other macroeconomic dynamics. General or industry specific market conditions or stock market performance or domestic or international macroeconomic and geopolitical factors unrelated to our performance also may affect the price of our stock.
In addition, the stock market in general, and the stocks of medical device companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. This could limit or prevent investors from readily selling their shares and may otherwise negatively affect the liquidity of our common stock. Securities class action litigation has often been instituted against companies following periods of volatility in the overall
56
market and in the market price of a company’s securities. This litigation, if instituted against us, could result in very substantial costs, divert our management’s attention and resources, and harm our business, financial condition and results of operation.
We expend substantial resources to comply with laws and regulations relating to public companies, and any failure to maintain compliance could subject us to regulatory scrutiny and cause investors to lose confidence in our company, which could harm our business and have a material adverse effect on our stock price.
Laws and regulations affecting public companies, including provisions of the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010 and the Sarbanes-Oxley Act of 2002, and the related rules and regulations adopted by the SEC, and by the Nasdaq Stock Market increase our accounting, legal and financial compliance costs and make some activities more time-consuming and costly. We cannot predict or estimate with any reasonable accuracy the total amount or timing of the costs we may incur to comply with these laws and regulations.
We are also subject to SEC regulations that require us to determine whether our products contain certain specified minerals, referred to under the regulations as “conflict minerals,” and, if so, to perform an extensive inquiry into our supply chain, to determine whether such conflict minerals originate from the Democratic Republic of Congo or an adjoining country. Compliance with these regulations has increased our costs and has been time-consuming for our management and our supply chain personnel (and time-consuming for our suppliers), and we expect that continued compliance will continue to require significant money and time. In addition, to the extent any of our disclosures are perceived by the market to be “negative,” it may cause customers to refuse to purchase our products. Further, if we determine to make any changes to products, processes, or sources of supply, it may result in additional costs, which may adversely affect our business, financial condition and results of operations.
Our amended and restated bylaws designates certain courts as the sole and exclusive forum for certain litigation that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us.
Our amended and restated bylaws provides that, unless we consent in writing to the selection of an alternative forum, to the fullest extent permitted by applicable law: (A) the Court of Chancery of the State of Delaware (or, if the Court of Chancery of the State of Delaware lacks subject matter jurisdiction, the Superior Court of the State of Delaware, or, if both the Court of Chancery of the State of Delaware and the Superior Court of the State of Delaware lack subject matter jurisdiction, the United States District Court for the District of Delaware) and any state (or, if applicable, federal) appellate court therefrom shall be the sole and exclusive forum for (i) any derivative action, suit, or proceeding brought on behalf of our company, (ii) any action, suit, or proceeding asserting a claim of breach of fiduciary duty owed by any current or former director, officer, or other employee, or stockholder of ours to our company or our stockholders or any action asserting a claim for aiding and abetting any such breach of fiduciary duty, (iii ) any action, suit, or proceeding asserting a claim against us or any of our directors, officers, or other employees arising pursuant to, or seeking to enforce any right, obligation, or remedy under, any provision of the General Corporation Law of Delaware (the “DGCL”) or our (certificate of incorporation or bylaws, (iv) any action, suit, or proceeding as to which the DGCL confers jurisdiction on the Court of Chancery of the State of Delaware, or (v) any action, suit, or proceeding asserting a claim against us or our current or former directors, officers, employees, or stockholders governed by the internal affairs doctrine, in all cases subject to the court’s having personal jurisdiction over the indispensable parties named as defendants (including personal jurisdiction by reason of any such indispensable party’s consent to personal jurisdiction in the State of Delaware or such court); and (B) the federal district courts of the United States shall be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act of 1933, as amended. These provisions may limit a stockholder’s ability to obtain a judicial forum that such stockholder may prefer for disputes governed by these provisions.
Environmental, social, and corporate governance (“ESG”) regulations, policies and provisions may make our supply chain more complex and may adversely affect our relationships with customers.
There is an increasing focus on the governance of environmental and social risks. A number of our customers who are payors or distributors have adopted, or may adopt, procurement policies that include ESG provisions that their suppliers or manufacturers must comply with, or they may seek to include such provisions in their terms and conditions. An increasing number of participants in the medical device industry are also joining voluntary ESG groups or organizations, such as the Responsible Business Alliance. These ESG provisions and initiatives are subject to change, can be unpredictable, and may be difficult and expensive for us to comply with, given the complexity of our supply chain and the outsourced manufacturing of certain components of our products. If we are unable to comply, or are unable to cause our suppliers to comply, with such policies or provisions, a customer may stop purchasing products from us, and may take legal action against us, which could harm our reputation, revenue, and results of operations.
57
Our business could be negatively impacted by corporate citizenship and ESG matters and/or our reporting of such matters.
There is an increasing focus from certain investors, customers, consumers, and other stakeholders concerning corporate citizenship and sustainability matters. We could be perceived as not acting responsibly in connection with these matters. Our business could be negatively impacted by such matters. Any such matters, or related corporate citizenship and sustainability matters, could have a material adverse effect on our business.
None.
Item 2. Properties
We lease or own real property to support our business. The following lists those properties that we believe are material to our business. We believe that our facilities meet our current needs and that we will be able to renew any such leases when needed on acceptable terms or find alternative facilities.
| | | | | | | | |
Facility | | Location | | Approx.
Square
Feet | | | Ownership |
Manufacturing, warehousing, distribution, research and development, location
of a cadaveric training laboratory, and administrative facility for Corporate
and allreporting segments | | Lewisville, TX | | | 140,000 | | | Leased |
Design, development, marketing, and inspection for biologics and spinal implant
products and distribution of certain spinal implant products; location of a
cadaveric training laboratory, and administrative facility | | Carlsbad, CA | | | 82,000 | | | Leased |
Manufacturing and distribution for certain Biologics products | | Irvine, CA | | | 70,000 | | | Leased |
Manufacturing, warehousing, distribution, research and development, and
administrative facility for Motion Preservation | | Sunnyvale, CA | | | 25,000 | | | Leased |
Design of Spinal Implants and location of a cadaveric training laboratory | | Wayne, PA | | | 3,700 | | | Leased |
Design, development, and marketing for Enabling Technologies products | | Toronto, CA | | | 9,200 | | | Leased |
Research and development, component manufacturing, quality control and
training facility for orthopedics products and sales management, distribution
and administrative facility for Italy | | Verona, Italy | | | 38,000 | | | Owned |
International distribution center for Orthofix products | | Verona, Italy | | | 18,000 | | | Leased |
Mechanical workshop for Orthofix products | | Verona, Italy | | | 9,000 | | | Leased |
Sales management, distribution and administrative facility for United Kingdom | | Maidenhead, England | | | 5,580 | | | Leased |
Sales management, distribution and administrative facility for Brazil | | São Paulo, Brazil | | | 22,000 | | | Leased |
Sales management, distribution and administrative facility for France | | Arcueil, France | | | 8,500 | | | Leased |
Sales management, distribution and administrative facility for Germany | | Ottobrunn, Germany | | | 18,300 | | | Leased |
Our manufacturing facilities are registered with the FDA. Our facilities are subject to FDA inspection to ensure compliance with its Quality System Regulations. For further information regarding the status of FDA inspections, see the "Item 1. Business - Government Regulation."
Item 3. Legal Proceedings
For a description of material pending legal proceedings, refer to Note 13 of the Notes to the Consolidated Financial Statements in Item 8 of this Annual Report.
Item 4. Mine Safety Disclosures
Not applicable.
58
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market for Our Common Stock
Our common stock is traded on the Nasdaq Global Select Market under the symbol “OFIX.” As of March 1, 2023, we had 493 holders of record of our common stock. A substantially greater number of holders of our common stock are “street name” or beneficial holders, whose shares are held by banks, brokers and other financial institutions. The closing price of our common stock on March 1, 2023 was $20.18. The following table shows the high and low sales prices for our common stock for each of the two most recent fiscal years.
| | | | | | | | |
| | High | | | Low | |
2021 | | | | | | |
First Quarter | | $ | 48.50 | | | $ | 39.34 | |
Second Quarter | | | 45.96 | | | | 39.23 | |
Third Quarter | | | 43.30 | | | | 36.35 | |
Fourth Quarter | | | 39.98 | | | | 28.65 | |
2022 | | | | | | |
First Quarter | | $ | 35.83 | | | $ | 29.75 | |
Second Quarter | | | 34.89 | | | | 23.54 | |
Third Quarter | | | 25.93 | | | | 19.11 | |
Fourth Quarter | | | 20.87 | | | | 14.33 | |
Dividends
We have not paid dividends to holders of our common stock in the past and have no present intention to pay dividends in the foreseeable future. Additionally, we have restrictions on our ability to pay dividends in certain circumstances pursuant to our Amended Credit Agreement. We currently intend to retain all of our consolidated earnings to finance the continued growth of our business.
In the event that we decide to pay a dividend to holders of our common stock in the future with dividends received from our subsidiaries, we may, based on prevailing rates of taxation, be required to pay additional withholding and income tax on such amounts.
Equity Compensation Plan Information
Information about our equity compensation plan is incorporated herein by reference to Part III, Item 12 of this report.
Recent Sales of Unregistered Securities
During the fourth quarter of 2022, we did not issue any securities that were not registered under the Securities Act of 1933, as amended (the "Securities Act").
Performance Graph
The following performance graph is not deemed to be “soliciting material” or to be “filed” with the SEC or subject to Regulation 14A or 14C or to the liabilities of Section 18 of the Exchange Act. This information will not be deemed to be incorporated by reference into any filing under the Securities Act of 1933 or the Exchange Act, except to the extent we specifically incorporate this information by reference.
59
The following graph compares our annual percentage change in cumulative total return on common shares over the past five years with the cumulative total return of companies comprising the NASDAQ Composite Index and the NASDAQ Stocks (SIC 3840-3849 US & Foreign) Surgical, Medical, and Dental Instruments and Supplies Index. This presentation assumes that $100 was invested in shares of the relevant issuers on December 31, 2017, and that dividends received were immediately invested in additional shares. The graph plots the value of the initial $100 investment at one-year intervals for the fiscal years shown. The NASDAQ Composite Index replaces the CRSP NASDAQ Stock Market (US and Foreign Companies) Index in this analysis and going forward, as the CRSP Index data is no longer accessible. The CRSP index has been included with data through 2022.
Item 6. Reserved
60
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and result of operations should be read in conjunction with “Forward-Looking Statements” and our consolidated financial statements and notes thereto appearing elsewhere in this Annual Report. The discussion and analysis below is focused on our 2022 and 2021 financial results, including comparisons of our year-over-year performance between these years. Discussion and analysis of our 2020 fiscal year specifically, as well as the year-over-year comparison of our 2021 financial performance to 2020, is located in Part II, Item 7 – Management’s Discussion and Analysis of Financial Condition and Results of Operations in our Annual Report on Form 10-K for the fiscal year ended December 31, 2021, filed with the SEC on February 25, 2022, which is available on our website at www.orthofix.com and the SEC’s website at www.sec.gov.
Merger with SeaSpine
On October 10, 2022, we entered into an Agreement and Plan of Merger with SeaSpine Holdings Corporation ("SeaSpine"), a global medical technology company focused on surgical solutions for the treatment of spinal disorders. On January 5, 2023, the transaction was completed, with SeaSpine continuing as a wholly-owned subsidiary of Orthofix following the transaction. As the merger was completed in 2023, SeaSpine's historical financial results for the year ended December 31, 2022, are not included within results of operations. As such, the information presented under the title "Results of Operations" below speaks only to the financial results of Orthofix on a stand-alone basis. However, the merger will have a significant impact on our future results of operations and financial condition. For example, the merger is expected to result in significant costs to achieve ongoing synergies, which could be associated with product line rationalization, employee severance and retention costs, professional fees for integration of processes and information technology systems, and other expenses. Future filings, beginning with our Quarterly Report on Form 10-Q for the fiscal quarter ending March 31, 2023, will reflect the results of the combined Orthofix-SeaSpine organization. For additional discussion related to the merger, see Note 22 of the Notes to the Consolidated Financial Statements in Item 8 of this Annual Report.
Executive Summary
The newly merged Orthofix-SeaSpine organization is a leading global spine and orthopedics company with a comprehensive portfolio of biologics, innovative spinal hardware, bone growth therapies, specialized orthopedic solutions, and a leading surgical navigation system. Its products are distributed in approximately 68 countries worldwide.
We are headquartered in Lewisville, Texas and have primary offices in Carlsbad, CA, with a focus on spine and biologics product innovation and surgeon education, and Verona, Italy, with an emphasis on product innovation, production, and medical education for orthopedics. Our combined global R&D, commercial and manufacturing footprint also includes facilities and offices in Irvine, CA, Toronto, Canada, Sunnyvale, CA, Wayne, PA, Olive Branch, MS, Maidenhead, UK, Munich, Germany, Paris, France and Sao Paulo, Brazil.
61
Notable financial results in 2022 include the following:
•Net sales were $460.7 million, a decrease of 0.8% on a reported basis and 1.5% on a constant currency basis
•Global Orthopedics net sales growth of 1.9% on a reported basis and 11.0% on a constant currency basis driven by strategic investments in our commercial channels and momentum from new product introductions
•FDA granted PMA approval for AccelStim LIPUS bone growth stimulator, expanding our indications into fresh fracture care
•Executed partnership with CGBio to commercialize Novosis rhBMP-2 growth factor in the U.S. and Canada
•Orthofix and MTF Biologics recognized with the 2022 Spine Technology Award from Orthopedics This Week for Virtuos Lyograft
Results of Operations
The following table presents certain items in our consolidated statements of operations as a percent of net sales:
| | | | | | | | | | | | |
| | Year ended December 31, | |
| | 2022
(%) | | | 2021
(%) | | | 2020
(%) | |
Net sales | | | 100.0 | | | | 100.0 | | | | 100.0 | |
Cost of sales | | | 26.8 | | | | 24.7 | | | | 25.1 | |
Gross profit | | | 73.2 | | | | 75.3 | | | | 74.9 | |
Sales and marketing | | | 49.7 | | | | 47.6 | | | | 50.3 | |
General and administrative | | | 17.4 | | | | 14.9 | | | | 16.7 | |
Research and development | | | 10.6 | | | | 10.7 | | | | 9.6 | |
Acquisition-related amortization and remeasurement | | | (1.6 | ) | | | 3.9 | | | | (0.2 | ) |
Operating income (loss) | | | (2.9 | ) | | | (1.8 | ) | | | (1.5 | ) |
Net income (loss) | | | (4.3 | ) | | | (8.3 | ) | | | 0.6 | |
Net Sales by Reporting Segment
The following table provides net sales by major product category by reporting segment:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Percentage Change | |
| | | | | | | | | | | 2022/2021 | | | 2022/2021 | | | 2021/2020 | | | 2021/2020 | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | | | Reported | | | Constant Currency | | | Reported | | | Constant Currency | |
Bone Growth Therapies | | $ | 187,247 | | | $ | 187,448 | | | $ | 171,396 | | | | -0.1 | % | | | -0.1 | % | | | 9.4 | % | | | 9.4 | % |
Spinal Implants | | | 109,546 | | | | 115,094 | | | | 94,857 | | | | -4.8 | % | | | -4.0 | % | | | 21.3 | % | | | 20.8 | % |
Biologics | | | 56,381 | | | | 56,421 | | | | 55,482 | | | | -0.1 | % | | | -0.1 | % | | | 1.7 | % | | | 1.7 | % |
Global Spine | | | 353,174 | | | | 358,963 | | | | 321,735 | | | | -1.6 | % | | | -1.4 | % | | | 11.6 | % | | | 11.4 | % |
Global Orthopedics | | | 107,539 | | | | 105,516 | | | | 84,827 | | | | 1.9 | % | | | 11.0 | % | | | 24.4 | % | | | 21.3 | % |
Net sales | | $ | 460,713 | | | $ | 464,479 | | | $ | 406,562 | | | | -0.8 | % | | | 1.5 | % | | | 14.2 | % | | | 13.5 | % |
62
Global Spine
Global Spine offers the following products categories:
-Bone Growth Therapies, which manufactures, distributes, sells, and provides support services for market leading devices that enhance bone fusion. Bone Growth Therapies uses distributors and sales representatives to sell its devices and provide associated services to hospitals, healthcare providers, and patients.
-Spinal Implants, which designs, develops, and markets a broad portfolio of motion preservation and fixation implant products used in surgical procedures of the spine. Spinal Implants distributes its products globally through a network of distributors and sales representatives to sell spine products to hospitals and healthcare providers.
-Biologics, which provides a portfolio of regenerative products and tissue forms that allow physicians to successfully treat a variety of spinal and orthopedic conditions. Biologics markets its tissues to hospitals and healthcare providers, primarily in the U.S., through a network of employed and independent sales representatives.
2022 Compared to 2021
Net sales decreased $5.8 million or 1.6%
•Bone Growth Therapies net sales were relatively flat, primarily driven by a continued slowdown in complex procedure volumes early in the year, which are typically paired with our CervicalStim and Spinalstim devices, largely offset by the successful commercial roll-out of our AccelStim Bone Healing Therapy in 2022 and growth in our PhysioStim product line from an expanding sales force and increased market share
•Spinal Implants net sales decreased $5.5 million or 4.8%, primarily due to lower complex procedures case volumes in Spine Fixation as well as global competitive pressures in Motion Preservation
•Biologics net sales were relatively flat, primarily driven by a decrease in volume from our cellular allograft offerings, which were largely offset by the impact of successful new product introductions, such as FiberFuse, FiberFuse Strip, Virtuos, and Legacy DBM
Global Orthopedics
Global Orthopedics offers products and solutions that allow physicians to successfully treat a variety of orthopedic conditions specifically related to limb reconstruction and deformity correction unrelated to the spine. Global Orthopedics distributes its products world-wide through a network of distributors and sales representatives to sell orthopedic products to hospitals and healthcare providers.
2022 Compared to 2021
Net sales increased $2.0 million, or 1.9% on a reported basis and 11.0% on a constant currency basis
•Double-digit growth internationally on a constant currency basis paired with solid growth in the U.S. from strategic investments in our commercial channels and momentum from new product introductions
•Partially offset by a decrease of $9.6 million due to movement in foreign currency exchange rates
Gross Profit
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Percentage Change | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | | | 2022/2021 | | | 2021/2020 | |
Net sales | | $ | 460,713 | | | $ | 464,479 | | | $ | 406,562 | | | | -0.8 | % | | | 14.2 | % |
Cost of sales | | | 123,544 | | | | 114,914 | | | | 101,889 | | | | 7.5 | % | | | 12.8 | % |
Gross profit | | $ | 337,169 | | | $ | 349,565 | | | $ | 304,673 | | | | -3.5 | % | | | 14.7 | % |
Gross margin | | | 73.2 | % | | | 75.3 | % | | | 74.9 | % | | | -2.1 | % | | | 0.4 | % |
2022 Compared to 2021
Gross profit decreased $12.4 million, or 3.5%
63
•Decrease in gross profit driven primarily by changes in our sales mix as well as increased inventory reserves related to set builds for an expanding sales force and increased safety stock requirements early in the year driven by the risk of global supply chain disruption
•Decrease also driven by unfavorable changes in shipping costs, increased manufacturing overhead costs, and unfavorable movements in foreign currency exchange rates
Sales and Marketing Expense
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Percentage Change | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | | | 2022/2021 | | | 2021/2020 | |
Sales and marketing | | $ | 228,810 | | | $ | 221,318 | | | $ | 204,434 | | | | 3.4 | % | | | 8.3 | % |
As a percentage of net sales | | | 49.7 | % | | | 47.6 | % | | | 50.3 | % | | | 2.1 | % | | | -2.7 | % |
2022 Compared to 2021
Sales and marketing expense increased $7.5 million
•Increases in travel, sales events, and surgeon and sales education trainings as in-person events largely resumed in 2022
•Increase also attributable to the hiring of additional sales and marketing headcount to support growth initiatives across both Spine and Orthopedics
•Increase of $2.4 million related to our estimated Italian Medical Device Payback liability, largely as a result of temporary relief provided by the Italian National Healthcare System in 2021 in response to the COVID-19 pandemic
General and Administrative Expense
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Percentage Change | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | | | 2022/2021 | | | 2021/2020 | |
General and administrative | | $ | 79,966 | | | $ | 69,353 | | | $ | 67,948 | | | | 15.3 | % | | | 2.1 | % |
As a percentage of net sales | | | 17.4 | % | | | 14.9 | % | | | 16.7 | % | | | 2.5 | % | | | -1.8 | % |
2022 Compared to 2021
General and administrative expense increased $10.6 million
•Increase of $12.0 million associated with due diligence, legal fees, and other acquisition-related costs incurred in order to close the merger with SeaSpine, and certain integration costs to prepare for the merging of activities on a combined company basis
•Partially offset by a decrease in certain compensation costs, partly stemming from the departure of certain former executives and from macroeconomic pressures on certain variable compensation expenses
Research and Development Expense
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Percentage Change | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | | | 2022/2021 | | | 2021/2020 | |
Research and development | | $ | 49,065 | | | $ | 49,621 | | | $ | 39,056 | | | | -1.1 | % | | | 27.1 | % |
As a percentage of net sales | | | 10.6 | % | | | 10.7 | % | | | 9.6 | % | | | -0.1 | % | | | 1.1 | % |
2022 Compared to 2021
Research and development expense decreased $0.6 million
•Decrease of $0.8 million related to the attainment of a development milestone with MTF Biologics achieved in 2021 that did not recur in 2022
•Decrease in integration activities attributable to certain recent asset acquisitions
•Partially offset by an increase of $2.3 million related directly to our European Union medical device regulation implementation efforts
64
Acquisition-related Amortization and Remeasurement
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Percentage Change | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | | | 2022/2021 | | | 2021/2020 | |
Acquisition-related amortization and remeasurement | | $ | (7,404 | ) | | $ | 17,588 | | | $ | (499 | ) | | | -142.1 | % | | | -3624.6 | % |
As a percentage of net sales | | | -1.6 | % | | | 3.9 | % | | | -0.2 | % | | | -5.5 | % | | | 4.1 | % |
2022 Compared to 2021
Acquisition-related amortization and remeasurement decreased $25.0 million
•Decrease of $14.0 million related to the remeasurement of potential revenue-based milestone payments associated with the Spinal Kinetics acquisition, as we do not expect to achieve the remaining revenue-based milestone prior to April 30, 2023, based on current net sales trends
•Decrease of $11.8 million attributable to the impairment of our Global Orthopedics goodwill in 2021
Non-operating Income (Expense)
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Percentage Change | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | | | 2022/2021 | | | 2021/2020 | |
Interest expense, net | | $ | (1,288 | ) | | $ | (1,837 | ) | | $ | (2,483 | ) | | | -29.9 | % | | | -26.0 | % |
Other income (expense) | | | (3,150 | ) | | | (3,343 | ) | | | 8,381 | | | | -5.8 | % | | | -139.9 | % |
Non-operating income and expense largely consists of interest income and expense, transaction gains and losses from changes in foreign currency exchange rates, changes in fair value related to our equity holdings in certain privately-held companies, and credit losses recognized on certain convertible debt investments. Foreign exchange gains and losses are primarily a result of several of our foreign subsidiaries holding trade and intercompany payables or receivables in currencies (most notably the U.S. Dollar) other than their functional currency.
2022 Compared to 2021
Interest expense, net, decreased $0.5 million
•Change primarily the result of increased interest income on money market funds as a result of increases in yield driven by domestic monetary policy
•Change also a result of increased interest income earned on certain investment securities
Other income (expense), net, increased $0.2 million
•Increase of $0.7 million associated with changes in foreign currency exchange rates, as we recorded a non-cash remeasurement loss of $3.3 million in 2022 compared to a loss of $4.0 million in 2021
•Partially offset by gains recognized in 2021 in total of $0.6 million associated with our equity investments in Neo Medical and Bone Biologics
Income Tax Expense
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Percentage Change | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | | | 2022/2021 | | | 2021/2020 | |
Income tax expense (benefit) | | $ | 2,043 | | | $ | 24,884 | | | $ | (2,885 | ) | | | -91.8 | % | | | -962.5 | % |
Effective tax rate | | | -11.5 | % | | | -184.4 | % | | | 784.0 | % | | | 172.9 | % | | | -968.4 | % |
2022 Compared to 2021
Net income tax expense decreased by $22.8 million
•Decrease of $20.2 million related to changes in valuation allowances recorded in 2021 versus 2022
•Decrease of $2.7 million related to the change in fair value of contingent consideration
•Partially offset by $1.0 million US tax expense on foreign income inclusion
65
A reconciliation of the effective tax rate for each year is reported in Note 20 to the Notes to the Consolidated Financial Statements contained in Item 8 of this Annual Report.
Segment Review
Historically, our business was managed through two reporting segments: Global Spine and Global Orthopedics. The primary metric used in managing the business by segment is EBITDA (which is described further in Note 16 to the Notes to the Consolidated Financial Statements contained in Item 8 of this Annual Report).
Following the merger with SeaSpine, which was completed on January 5, 2023, we expect to reassess our reporting segments in the first quarter of 2023 based on how the operations of the newly combined company will be managed. We will also reassess our identified segment profitability metric at that time. Accordingly, the reporting segment information below has been prepared based on our two historical reporting segments, which were utilized in managing operations for the year ended December 31, 2022.
The following table reconciles EBITDA to loss before income taxes:
| | | | | | | | | | | | |
| | Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Global Spine | | $ | 60,649 | | | $ | 58,014 | | | $ | 63,036 | |
Global Orthopedics | | | (4,037 | ) | | | 3,374 | | | | (4,993 | ) |
Corporate | | | (44,011 | ) | | | (31,691 | ) | | | (25,382 | ) |
Total EBITDA | | | 12,601 | | | | 29,697 | | | | 32,661 | |
Depreciation and amortization | | | (29,019 | ) | | | (29,599 | ) | | | (30,546 | ) |
Goodwill impairment | | | — | | | | (11,756 | ) | | | — | |
Interest expense, net | | | (1,288 | ) | | | (1,837 | ) | | | (2,483 | ) |
Loss before income taxes | | $ | (17,706 | ) | | $ | (13,495 | ) | | $ | (368 | ) |
Liquidity and Capital Resources
Cash, cash equivalents, and restricted cash at December 31, 2022, was $50.7 million compared to $87.8 million at December 31, 2021.
| | | | | | | | | | | | |
| | Year Ended December, 31, | | | | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | Change | |
Net cash from operating activities | | $ | (11,538 | ) | | $ | 18,475 | | | $ | (30,013 | ) |
Net cash from investing activities | | | (24,534 | ) | | | (23,013 | ) | | | (1,521 | ) |
Net cash from financing activities | | | (78 | ) | | | (3,621 | ) | | | 3,543 | |
Effect of exchange rate changes on cash and restricted cash | | | (997 | ) | | | (815 | ) | | | (182 | ) |
Net change in cash, cash equivalents, and restricted cash | | $ | (37,147 | ) | | $ | (8,974 | ) | | $ | (28,173 | ) |
The following table presents free cash flow, a non-GAAP financial measure, which is calculated by subtracting capital expenditures from net cash from operating activities.
| | | | | | | | | | | | |
| | Year Ended December, 31, | | | | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | Change | |
Net cash from operating activities | | $ | (11,538 | ) | | $ | 18,475 | | | $ | (30,013 | ) |
Capital expenditures | | | (23,160 | ) | | | (19,592 | ) | | | (3,568 | ) |
Free cash flow | | $ | (34,698 | ) | | $ | (1,117 | ) | | $ | (33,581 | ) |
Operating Activities
Cash flows from operating activities decreased $30.0 million
•Decrease in net loss of $18.6 million
•Net decrease of $44.1 million in non-cash gains and losses, largely related to our impairment of Global Orthopedics goodwill in 2021, changes in deferred income taxes, and changes in fair value of contingent consideration
66
•Net decrease of $4.5 million relating to changes in working capital accounts, primarily attributable to changes in inventory levels, our contract liability associated with the CMS Accelerated and Advance Payment Program, the payment of contingent consideration in 2021, and from changes in other long-term assets and liabilities
Two of our primary working capital accounts are accounts receivable and inventory. Day’s sales in receivables were 62 days at December 31, 2022, compared to 58 days at December 31, 2021 (calculated using fourth quarter net sales and ending accounts receivable). Inventory turns were 1.2 times as of December 31, 2022, compared to 1.4 times at December 31, 2021.
Investing Activities
Cash flows from investing activities decreased $1.5 million
•Decrease of $3.6 million associated with capital expenditures compared to the prior year period
•Partially offset by an increase of $2.2million due to cash paid for purchases of investment securities in 2021
Financing Activities
Cash flows from financing activities increased $3.5 million
•Increase of $8.4 million associated with cash paid in 2021 for the achievement of a revenue-based milestone associated with the Spinal Kinetics acquisition; the milestone payment totaled $15.0 million with a portion of the payment reflected in both operating and financing activities
•Decrease in net proceeds of $3.7 million from the issuance of common shares, primarily related to the exercise of stock options in the prior year period
•Decrease of $2.0 million related to the conclusion of the FITBONE Contract Manufacturing and Supply Agreement with Wittenstein, resulting in a $2.0 million payment in 2022
•Increase of $0.9 million attributable to other financing activities
Credit Facilities
On October 25, 2019, we entered into a Second Amended and Restated Credit Agreement (the “Amended Credit Agreement”), which provides for a five year $300 million secured revolving credit facility. The Amended Credit Agreement has a maturity date of October 25, 2024, and amends and restates the previous $125 million secured revolving credit facility.
Borrowings under the Amended Credit Agreement may be used for, among other things, working capital and other general corporate purposes (including share repurchases, permitted acquisitions and permitted payments of dividends and other distributions). Borrowings under the Amended Credit Agreement may be limited based on EBITDA levels recognized over the preceding 12 months.
As of December 31, 2022, we have no outstanding borrowings under the Amended Credit Agreement. However, on January 3, 2023, we borrowed $30.0 million under the $300.0 million secured revolving credit facility for working capital purposes, including to fund certain merger-related expenses. Further, an additional $15.0 million was borrowed on March 3, 2023. For additional information regarding the credit facility, see Note 11 of the Notes to the Consolidated Financial Statements in Item 8 of this Annual Report.
In addition, we have no outstanding borrowings on our Italian line of credit of €5.5 million ($6.3 million) as of December 31, 2022. This unsecured line of credit provides us the option to borrow amounts in Italy at rates which are determined at the time of borrowing.
As of December 31, 2022, SeaSpine had a $30.0 million credit facility with Wells Fargo Bank, National Association which was scheduled to mature in July 2025. Immediately prior to the closing date of the merger, SeaSpine had $27.0 million of outstanding borrowings under the credit facility. In connection with the merger, on January 5, 2023, all of the outstanding obligations in respect of principal, interest, and fees under the credit agreement were repaid and all applicable commitments under the credit agreement were terminated.
Other
For information regarding Contingencies, see Note 13 of the Notes to the Consolidated Financial Statements in Item 8 of this Annual Report.
67
Legion Innovations, LLC Asset Acquisition
On December 29, 2022, we entered into a technology assignment and royalty agreement with Legion Innovations, LLC, a U.S.-based medical device technology company, whereby we acquired intellectual property rights to certain assets. As consideration, we paid $0.2 million in January 2023, with additional payments contingent upon reaching future commercialization and revenue-based milestones.
CGBio Co. Ltd. License and Distribution Agreement
On July 30, 2022, we entered into an exclusive License and Distribution Agreement with CGBio Co., Ltd. (“CGBio”), a developer of innovative, synthetic bone grafts. The agreement grants us the exclusive right to conduct pre-clinical and clinical studies, commercialize, promote, market, and sell the Novosis recombinant human bone morphogenetic protein-2 (rhBMP-2) bone growth materials and other future tissue regenerative solutions in the U.S. and Canada. As consideration, we paid CGBio an upfront payment of $1.4 million with additional payments contingent upon the achievement of specified development milestones.
Spinal Kinetics Acquisition and Contingent Consideration
As part of the consideration for the Spinal Kinetics acquisition, we agreed to make contingent milestone payments of up to $60.0 million. One milestone payment, which was for $15.0 million, became due upon FDA approval of Spinal Kinetics’ M6-C artificial cervical disc, which was achieved and paid in 2019. A revenue-based milestone payment, totaling $15.0 million, was achieved and paid in 2021 upon meeting certain net sales targets.
The remaining milestone payment is a revenue-based milestone payment of $30.0 million in connection with future sales of the acquired artificial discs. The fair value of this contingent consideration liability was concluded to be zero as of December 31, 2022, as we do not expect to achieve the milestone prior to the deadline of April 30, 2023. For additional discussion of this matter, see Note 12 of the Notes to the Consolidated Financial Statements in Item 8 of this Annual Report.
IGEA S.p.A Exclusive License and Distribution Agreement
In April 2021, we entered into an Exclusive License and Distribution Agreement (the “License Agreement”) with IGEA S.p.A (“IGEA”), an Italian manufacturer and distributor of bone and cartilage stimulation systems. Per the terms of the License Agreement, we have the exclusive right to sell IGEA products in the U.S. and Canada. As consideration for the License Agreement, we agreed to pay up to $4.0 million, of which $0.5 million was paid in 2021, with certain payments contingent upon achieving an FDA milestone.
In May 2022, the Company achieved FDA approval pertaining to the acquired technology, triggering a contingent consideration milestone obligation of $3.5 million. Of this amount, $1.5 million was paid in 2022, $1.0 million was accrued within other current liabilities, and $1.0 million was accrued within other long-term liabilities as of December 31, 2022.
Related Party Transaction
In February 2021, we entered into a technology assignment and royalty agreement with a medical device technology company partially owned and controlled by the wife of our Executive Chairman, and former President and Chief Executive Officer, Jon Serbousek, whereby we acquired the intellectual property rights to certain assets for consideration of up to $10.0 million. Consideration was comprised of $1.0 million, which was paid at signing, and $9.0 million in contingent consideration, dependent upon multiple milestones, such as receipt of 510(k) clearance or the attainment of certain net sales targets. For additional discussion of this transaction, see Note 17 of the Notes to the Consolidated Financial Statements in Item 8 of this Annual Report.
Neo Medical Investment and Convertible Loan
In October 2020, we entered into a Convertible Loan Agreement (the “Convertible Loan”) with Neo Medical SA, a privately held Swiss-based Medtech company (“Neo Medical”), whereby we loaned CHF 4.6 million to Neo Medical ($5.0 million as of the issuance date). The loan bears interest at 8.0%, with interest due semi-annually. The Convertible Loan matures in October 2024; however, if a change in control of Neo Medical occurs prior to maturity, the Convertible Loan shall become immediately due upon such event.
Impact of COVID-19 and the Coronavirus Aid, Relief, and Economic Security Act ("CARES Act") on Liquidity and Capital Resources
In April 2020, we received $13.9 million in funds from the CMS Accelerated and Advance Payment Program as part of the CARES Act to increase cash flow to providers of services and suppliers impacted by the COVID-19 pandemic. In April 2021, Medicare began to recoup 25% of Medicare payments otherwise owed to the provider or supplier for submitted claims. Recoupment then increased to 50% of Medicare payments in March 2022. Thus, during these time periods, rather than receiving the full amount of payment for
68
newly submitted claims, our outstanding balance under the Accelerated and Advance Payment Program was reduced by the recoupment amount until the full balance had been repaid, which was completed in 2022.
Disclosures Relating to Potential Obligations Resulting from our Merger with SeaSpine
On December 1, 2022, SeaSpine entered into an exclusive license and distribution agreement with Lattus Spine LLC. (“Lattus”) to acquire a perpetual license to certain surgical instrumentation. As consideration, SeaSpine paid an upfront licensing fee and purchased initial sets of instrumentation. Additional payments are contingent upon the subsequent net sales of the acquired assets, most of which are based upon future net sales of the acquired assets, with the company having the option to fund a portion of these payments via the issuance of common stock.
Pursuant to a distributor agreement between SeaSpine and one of its distributors, the Company could be obligated to purchase certain assets of the distributor, at the option of the distributor, as a result of the merger. If this were to occur, the purchase price of the transaction would be the greater of (i) $4.2 million or (ii) 100% of the commissions earned by the distributor over a specified period prior to the change in control, with such purchase price paid in stock.
Unremitted Foreign Earnings
Unremitted foreign earnings were $27.0 million as of December 31, 2022. The Company’s investment in foreign subsidiaries continues to be indefinite in nature; however, the Company may periodically repatriate a portion of these earnings to the extent that it does not incur significant additional tax liability.
Contractual Obligations
As a result of our operations, we are subject to certain contractual obligations with material cash requirements. Our material contractual obligations include, but are not limited to i) our contingent consideration arrangement associated with the Spinal Kinetics acquisition, ii) contingent consideration arrangements associated with certain asset acquisitions, of which material obligations are described above, iii) operating lease and finance lease obligations, and iv) uncertain tax positions.
Refer to the Notes to the Consolidated Financial Statements in Item 8 of this Annual Report for a further description of our contingent consideration arrangements (Notes 12 and 17), lease obligations (Note 9), and uncertain tax positions (Note 20).
Off-balance Sheet Arrangements
As of December 31, 2022, we did not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, cash flows, liquidity, capital expenditures, or capital resources that are material to investors. In addition, we do not consider the backlog of firm orders to be material.
Critical Accounting Estimates
Our discussion of operating results is based upon the consolidated financial statements and accompanying notes. The preparation of these statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities as of the date of the financial statements and the reported amount of revenues and expenses during the reporting period. On an ongoing basis, we evaluate these estimates, which are based on historical experience and various other assumptions that management believe to be reasonable under the circumstances at that point in time. Actual results may differ, significantly at times, from these estimates.
We believe the estimates described below are the most critical in preparing our consolidated financial statements. We have reviewed these critical accounting estimates with the Audit Committee of the Board of Directors.
Revenue Recognition
The process for recognizing revenue involves significant assumptions and judgments for certain of our revenue streams. Revenue recognition policies are “critical accounting estimates” because changes in the assumptions used to develop the estimates could materially affect key financial measures, including net sales, gross margin, operating income, EBITDA, and net income.
Bone Growth Therapies revenue is largely attributable to the U.S. and is comprised of third-party payor transactions and wholesale revenue.
69
For revenue derived from third-party payors, including commercial insurance carriers, health maintenance organizations, preferred provider organizations, and governmental payors, such as Medicare, in connection with the sale of our stimulation products, we recognize revenue when the stimulation product is fitted to and accepted by the patient and all applicable documents that are required by the third-party payor have been obtained. Amounts paid by these third-party payors are generally based on fixed or allowable reimbursement rates. These revenues are recorded at the expected or preauthorized reimbursement rates, net of any contractual allowances or adjustments. Certain billings are subject to review by the third-party payors and may be subject to adjustment.
Wholesale revenue is related to the sale of our bone growth stimulators directly to physicians and other healthcare providers. Wholesale revenues are recognized upon shipment and receipt of a confirming purchase order, which is when the customer obtains control of the promised goods.
Biologics revenue is largely attributable to the U.S. and is primarily related to a collaborative arrangement with MTF. We have exclusive global marketing rights and receive marketing fees from MTF based on products distributed by MTF. MTF is considered the principal in these arrangements; therefore, we recognize these marketing service fees on a net basis upon shipment of the product to the customer and receipt of a confirming purchase order.
Spinal Implants and Global Orthopedics products are distributed world-wide, with U.S. sales largely comprised of commercial revenue and international sales derived from commercial sales and through stocking distributor arrangements.
Commercial revenue is largely related to the sale of our Spinal Implants and Global Orthopedics products to hospital customers. Commercial revenues are recognized when these products have been utilized and a confirming purchase order has been received from the hospital.
Stocking distributors purchase our products and then re-sell them directly to customers, such as hospitals. Revenue derived from stocking distributor arrangements is recognized upon shipment and receipt of a confirming purchase order, which is when the distributor obtains control of the promised goods. The transaction price is estimated based upon our historical collection experience with the stocking distributor. This percentage, which is specific to each stocking distributor, is then used to calculate the transaction price. Cost of sales is also recorded upon transfer of control of the product to the customer, which is when our performance obligation has been satisfied.
Allowance for Expected Credit Losses and Contractual Allowances
The process for estimating the ultimate collection of accounts receivable involves significant assumptions and judgments. The determination of the contractual life of accounts receivable, the aging of outstanding receivables, as well as the historical collections, write-offs, and payor reimbursement experience over the estimated contractual lives of such receivables, are integral parts of the estimation process related to reserves for expected credit losses and the establishment of contractual allowances. Accounts receivable are analyzed on a quarterly basis to assess the adequacy of both reserves for expected credit losses and contractual allowances. Revisions in allowances for expected credit loss estimates are recorded as an adjustment to bad debt expense within sales and marketing expenses. Revisions to contractual allowances are recorded as an adjustment to net sales. These estimates are periodically tested against actual collection experience. In addition, we analyze our receivables by geography and by customer type, where appropriate, in developing estimates for expected credit losses.
We believe our allowance for credit losses is sufficient to cover customer credit risks; however, a 10% change in our allowance for credit losses as of December 31, 2022, would result in an increase or decrease to sales and marketing expense of $0.6 million. Additionally, we believe our estimate to establish contractual allowances is sufficient to cover customer credit risks; however, a 10% change in our reserve for contractual allowances as of December 31, 2022, would result in an increase or decrease to net sales of $0.3 million. Our allowance for credit losses and estimation of contractual allowances are “critical accounting estimates” because changes in the assumptions used to develop the estimates could materially affect key financial measures, including net sales, gross margin, operating income, EBITDA, net income, and accounts receivable.
Inventory Allowances
Reserves for excess, slow moving, and obsolete inventory are calculated as the difference between the cost of inventory and market value, and are based on assumptions and judgments about new product launch periods, overall product life cycles, forecasted demand, and market conditions. In the event of a decrease in demand for our products, excess product production, or a higher
70
incidence of inventory obsolescence, we could be required to increase our inventory reserves, which would increase cost of sales and decrease gross profit. We regularly evaluate our exposure for inventory write-downs. If conditions or assumptions used in determining the market value or forecasted demand change, additional inventory adjustments in the future may be necessary. Our inventory allowance is a “critical accounting estimate” because changes in the assumptions used to develop the estimate could materially affect key financial measures, including gross profit, operating income, EBITDA, net income, and inventory.
Valuation of Intangible Assets
Our intangible assets are comprised primarily of patents, acquired or developed technology, in-process research and development (“IPR&D”), customer relationships, trade names, trademarks, and licensing arrangements. We make significant judgments in relation to the valuation of intangible assets resulting from business combinations or asset acquisitions. Intangible assets acquired in a business combination that are used for IPR&D activities are considered to have indefinite lives until the completion or abandonment of the associated project. Upon reaching the end of the relevant project, we will either amortize the acquired IPR&D over its estimated useful life or expense the acquired IPR&D should the project be unsuccessful with no future alternative use.
Significant judgment is required related to the forecasting of future operating results within our discounted cash flow valuation models to determine the valuation of intangible assets. Key assumptions include the anticipated useful lives of acquired intangibles, the projected cash flows associated with each intangible asset, the estimated probability of success for acquired IPR&D projects, and projected growth rates and discount rates. It is possible that significant changes in plans or assumptions may affect the recoverability of these assets and could potentially result in impairment. Our valuation of intangible assets is a “critical accounting estimate” because changes in the assumptions used to develop these estimates could materially affect key financial measures, including operating income, EBITDA, and net income.
Goodwill
Our goodwill represents the excess of cost over fair value of net assets acquired from business combinations. The determination of the value of goodwill and intangible assets arising from business combinations requires extensive use of accounting estimates and judgments to allocate the purchase price to the fair value of the net tangible and intangible assets acquired.
We test goodwill at least annually for impairment, and between annual tests if indicators of potential impairment exist. These indicators include, among others, significant declines in sales, earnings, or cash flows, or the development of a material adverse change in the business climate. Assessing goodwill impairment involves a high degree of judgment due to the estimates and assumptions used. We believe the estimates and assumptions involved in the impairment assessment to be critical because significant changes in such estimates and assumptions could materially affect key financial measures, including operating income, EBITDA, and net income.
In the fourth quarter of 2021, we performed a quantitative assessment of goodwill as part of our annual goodwill impairment analysis. Upon estimating the fair value of each of its reporting units, we determined the Global Orthopedics reporting unit’s fair value was less than its carrying value of net assets. This resulted in recording a full impairment of the Global Orthopedics goodwill of $11.8 million, which is reflected within Acquisition-related amortization and remeasurement. The assessment concluded there were no indicators of impairment for the Global Spine goodwill.
In the fourth quarter of 2022, we performed a qualitative assessment for our annual goodwill impairment analysis, which did not result in an impairment charge. This qualitative analysis considered all relevant factors specific to the reporting units, including macroeconomic conditions, industry and market considerations, overall financial performance, and relevant entity-specific events. As part of our qualitative assessment, we included quantitative factors to assess the likelihood of an impairment and concluded it more likely that not that an impairment has not occurred.
We estimate the fair value of each reporting unit using a weighted average of fair value derived from both an income approach and a market approach. The fair value measurements are based on significant inputs that are unobservable in the market, with key assumptions including, but not limited to, our forecasted future net sales and expenses, terminal growth rates, discount rates applied, and allocation of corporate-level expenses to each reporting unit. Significant changes in these assumptions could result in a significantly higher or lower fair value, which in turn can affect the ultimate conclusion regarding if goodwill is impaired.
Fair Value Measurements
71
Fair value is defined as the price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. The two most significant items that are or were recorded at fair value as of December 31, 2022, and 2021, include (i) contingent consideration attributable to the Spinal Kinetics acquisition and (ii) our convertible loan agreements with Neo Medical.
The contingent consideration consists of potential future milestone payments of up to $60.0 million in cash associated with the Spinal Kinetics acquisition, which must be achieved before April 30, 2023, to be paid. The milestone payments include (i) up to $15.0 million for meeting the FDA Milestone and (ii) revenue-based milestone payments of up to $45.0 million in connection with future sales of the M6-C artificial cervical disc and the M6-L artificial lumbar disc. The FDA milestone was achieved and paid in 2019 and one of the revenue-based milestones, resulting in a payment of $15.0 million, was achieved and paid in 2021.
As of December 31, 2022, we estimated the fair value of the remaining revenue-based milestone based on a probability-weighted cash flow analysis, based upon a combination of our historical net sales of artificial cervical discs and projected net sales of such discs through April 30, 2023. The estimated fair value of the remaining milestone was concluded to be zero as of December 31, 2022, as we considered the probability of achieving the milestone by April 30, 2023, to be remote.
In previous periods, we estimated the fair value of the remaining revenue-based milestone payment using a Monte Carlo simulation. This fair value measurement was based on significant inputs that were unobservable in the market, with key assumptions including our forecasted future net sales of Motion Preservation products, discount rates applied, and assumptions for potential volatility of the forecasted revenue. Significant changes in these assumptions could have resulted in a significantly higher or lower fair value as of each period.
We estimate the fair value of our convertible loan agreements with Neo Medical using option-pricing models and a probability-weighted discounted cash flow model. The fair value measurement is based on significant inputs that are unobservable in the market, with significant unobservable inputs including applicable discount rates, implied volatility, the likelihood and projected timing of repayment or conversion, and projected cash flows in support of the estimated enterprise value of Neo Medical. Significant changes in these assumptions could result in a significantly higher or lower fair value. Holding other inputs constant, an increase in the assumed cost of equity discount rate by 2% would have resulted in a decrease in the fair value of the convertible loan of $0.1 million, whereas a decrease the cost of equity discount rate by 2% would have resulted in an increase in the fair value of the convertible loan by $0.2 million.
Our fair value measurements are a “critical accounting estimate” because changes in the assumptions used to develop the estimate could materially affect key financial measures, including operating income, EBITDA, and net income.
Litigation and Contingent Liabilities
From time to time, we are parties to or targets of lawsuits, investigations, and proceedings, including product liability, personal injury, patent and intellectual property, health and safety, and employment and healthcare regulatory matters, which are handled and defended in the ordinary course of business. These lawsuits, investigations, or proceedings could involve a substantial number of claims and could also have an adverse impact on our reputation and customer base. Although we maintain various liability insurance programs for liabilities that could result from such lawsuits, investigations, or proceedings, we are self-insured for a significant portion of such liabilities.
We accrue for such claims when it is probable that a liability has been incurred and the amount can be reasonably estimated. The assessments of whether a loss is probable or a reasonable possibility, and whether the loss or range of loss is reasonably estimable, often involve a series of complex judgments about future events. Among the factors that we consider in this assessment are the nature of existing legal proceedings, investigations, and claims, the asserted or possible damages or loss contingency (if reasonably estimable), the progress of the matter, existing law and precedent, the opinions or views of legal counsel and other advisers, the involvement of the U.S. Government and its agencies in such proceedings, our experience in similar matters and the experience of other companies, the facts available to us at the time of assessment, and how we intend to respond, or have responded, to the proceeding, investigation or claim. Our assessment of these factors may change over time as individual proceedings, investigations or claims progress. For matters where we are not currently able to reasonably estimate the range of reasonably possible loss, the factors that have contributed to this determination include the following: (i) the damages sought are indeterminate, or an investigation has not manifested itself in a filed civil or criminal complaint, (ii) the matters are in the early stages, (iii) the matters involve novel or unsettled legal theories or a large or uncertain number of actual or potential cases or parties, and/or (iv) discussions with the government or other parties in matters that may be expected ultimately to be resolved through negotiation and settlement have not reached the point where we believe a reasonable estimate of loss, or range of loss, can be made. In such instances, we
72
believe that there is considerable uncertainty regarding the timing or ultimate resolution of such matters, including a possible eventual loss, fine, penalty or business impact, if any.
Changes in the facts and circumstances associated with a claim could have a material impact on our results of operations and cash flows in the period that reserve estimates are recorded or revised. We believe our insurance coverage and reserves are sufficient to cover currently estimated exposures, but we cannot give any assurance that we will not incur liabilities in excess of recorded reserves or our present insurance coverage. Litigation and contingent liabilities are “critical accounting estimates” because changes in the assumptions used to develop the estimates could materially affect key financial measures, including operating income, EBITDA, and net income.
Tax Matters
We and each of our subsidiaries are taxed at the rates applicable within each of their respective jurisdictions. Our income tax expense, effective tax rate, deferred tax assets, and deferred tax liabilities will vary according to the jurisdiction in which profits arise. Further, certain of our subsidiaries sell products directly to our other subsidiaries or provide administrative, marketing, and support services to our other subsidiaries. These intercompany sales and support services involve subsidiaries operating in jurisdictions with differing tax rates. The tax authorities in such jurisdictions may challenge our treatment under residency criteria, transfer pricing provisions, or other aspects of their respective tax laws, which could affect our composite tax rate and provisions.
We sometimes engage in transactions in which tax consequences may be subject to uncertainty. We account for these uncertain tax positions in accordance with applicable accounting guidance, which requires significant judgment in assessing the estimated tax consequences of a transaction. We evaluate the tax position taken or expected to be taken in a tax return by determining if the weight of available evidence indicates that it is more likely than not that, on an evaluation of the technical merits, the tax position will be sustained on audit, including resolution of any related appeals or litigation processes. We measure the tax benefit as the largest amount that is more than 50% likely to be realized upon ultimate settlement. We re-evaluate our income tax positions periodically to consider factors such as changes in facts or circumstances, changes in or interpretations of tax law, effectively settled issues under audit, and new audit activity. Such a change in recognition or measurement would result in recognition of a tax benefit or an additional charge to the tax provision, which could have a material impact to the financial statements.
We establish a valuation allowance when measuring deferred tax assets if it is more likely than not that certain deferred tax assets will not be realized in the foreseeable future. This process requires significant judgment as we must project the current tax liability and estimate the deferred tax assets and liabilities into future periods, including net operating loss and tax credit carry forwards. In assessing the need for a valuation allowance, we consider recent operating results, availability of taxable income in carryback years, future reversals of taxable temporary differences, future taxable income projections (exclusive of reversing temporary differences), and all prudent and feasible tax planning strategies.
Tax matters are “critical accounting estimates” because changes in the assumptions used to develop the estimates could materially affect key financial measures, including net income.
Share-based compensation
We use the Black-Scholes valuation model to calculate the fair value of service-based stock options. The value is recognized as expense over the service period net of actual forfeitures. The expected term of options granted is estimated based on a number of factors, including the vesting and expiration terms of the award, historical employee exercise behavior for both options that are currently outstanding and options that have been exercised or are expired, the historical volatility of our common stock, and an employee’s average length of service. The risk-free interest rate is determined based upon a constant U.S. Treasury security rate with a contractual life that approximates the expected term of the option award. We estimate expected volatility based on the historical volatility of our stock.
We use the Monte Carlo valuation methodology to calculate the fair value of market-based restricted stock units. The value is recognized as expense over the requisite service period and adjusted for forfeitures as they occur. The Monte Carlo methodology that we use to estimate the fair value of the awards incorporates the possibility that the market condition may not be satisfied.
The fair value of performance-based restricted stock units is calculated based upon (i) the closing stock price at the date of grant and (ii) the number of stock units expected to vest at the conclusion of the performance period. The value is recognized as expense over
73
the derived requisite service period beginning in the period in which the grants are deemed probable to vest. Vesting probability is assessed based upon forecasted financial results and requires significant judgment.
Determining the appropriate fair value model and calculating the fair value of employee stock awards requires estimates and judgments. Our share-based compensation is a “critical accounting estimate” because changes in the assumptions used to develop estimates of fair value or the requisite service period could materially affect key financial measures, including gross profit, operating income, EBITDA, and net income.
Non-GAAP Financial Measures
We believe that providing non-GAAP financial measures that exclude certain items provides investors with greater transparency to the information used by senior management in its financial and operational decision-making. We believe it is important to provide investors with the same non-GAAP metrics that senior management uses to supplement information regarding the performance and underlying trends of our business operations in order to facilitate comparisons to historical operating results and internally evaluate the effectiveness of our operating strategies. Disclosure of these non-GAAP financial measures also facilitates comparisons of our underlying operating performance with other companies in the industry that also supplement their GAAP results with non-GAAP financial measures.
The non-GAAP financial measures used in this Annual Report may have limitations as analytical tools and should not be considered in isolation or as a replacement for GAAP financial measures. Some of the limitations associated with the use of these non-GAAP financial measures are that they exclude items that reflect an economic cost that can have a material effect on cash flows. Similarly, certain non-cash expenses, such as equity compensation expense, do not directly impact cash flows, but are part of total compensation costs accounted for under GAAP.
Constant Currency
Constant currency is a non-GAAP measure, which is calculated by using foreign currency rates from the comparable, prior-year period, to present net sales at comparable rates. Constant currency can be presented for numerous GAAP measures, but is most commonly used by management to analyze net sales without the impact of changes in foreign currency rates.
EBITDA
EBITDA is defined as earnings before interest income (expense), net, income taxes, depreciation, and amortization (including the impacts of any goodwill impairment). EBITDA is the primary metric used by our Chief Operating Decision Maker in managing the business.
Free Cash Flow
Free cash flow is a non-GAAP financial measure, which is calculated by subtracting capital expenditures from net cash from operating activities. Free cash flow is an important indicator of how much cash is generated or used by our normal business operations, including capital expenditures. Management uses free cash flow as a measure of progress on its capital efficiency and cash flow initiatives.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
We are exposed to certain market risks as part of our ongoing business operations. Primary exposures include changes in interest rates and foreign currency fluctuations. These exposures can impact sales, cost of sales, costs of operations, and the cost of financing and yields on cash and short-term investments. We may use derivative financial instruments, where appropriate, to manage these risks. However, our risk management policy does not allow us to hedge positions we do not hold nor do we enter into derivative or other financial investments for trading or speculative purposes.
We are exposed to interest rate risk in connection with our Revolving Credit Facility, which bears interest at floating rates based on the Secured Overnight Financing Rate, or SOFR, plus an applicable borrowing margin or at a base rate (as defined in the Amended Credit Agreement) plus an applicable borrowing margin. Therefore, interest rate changes generally do not affect the fair market value of the debt, but do impact future earnings and cash flows, assuming other factors are held constant. As we do not have any balance outstanding associated with the Amended Credit Agreement as of December 31, 2022, this risk is currently minimal.
74
However, on January 3, 2023, we borrowed $30.0 million under Revolving Credit Facility for working capital purposes, including to fund certain merger-related expenses. Further, an additional $15.0 million was borrowed on March 3, 2023.
We believe that a concentration of credit risk related to our accounts receivable is limited because our customers are geographically dispersed and the end users are diversified across several industries. It is reasonably possible that changes in global economic conditions and/or local operating and economic conditions in the regions these customers operate, or other factors, could affect the future realization of these accounts receivable balances.
Our foreign currency exposure results from fluctuating currency exchange rates, primarily the U.S. Dollar against the Euro, Brazilian Real, Australian Dollar, Swiss Franc, or British Pound. We are subject to transactional currency exposures when our subsidiaries (or the Company itself) enter into transactions denominated in a currency other than their functional currency. For the year ended December 31, 2022, we recorded a foreign currency loss of $3.3 million on the statement of operations and comprehensive income (loss) resulting from gains and losses in foreign currency transactions.
We are also subject to currency exposure from translating the results of our global operations into the U.S. Dollar at exchange rates that fluctuate during the period. The U.S. Dollar equivalent of international sales denominated in foreign currencies was unfavorably impacted during the year ended December 31, 2022, and favorably impacted during the year ended December 31, 2021, by monthly foreign currency exchange rate fluctuations of the U.S. Dollar against all of the foreign functional currencies for our international operations. As we continue to distribute and manufacture our products in selected foreign countries, we expect that future sales and costs associated with our activities in these markets will continue to be denominated in the applicable foreign currencies, which could cause currency fluctuations to materially impact our operating results. An analysis was performed to determine the sensitivity of our current year net sales and operating income to changes in foreign currency exchange rates. We determined that if the U.S. Dollar decreased in value by 10% relative to all foreign currencies of our international operations it would result in an increase in net sales of $9.0 million and an increase in operating income of $0.8 million. If the U.S. Dollar increased in value by 10% relative to all foreign currencies of our international operations it would result in a decrease in net sales of $9.0 million and a decrease in operating income of $0.8 million.
Item 8. Financial Statements and Supplementary Data
See “Index to Consolidated Financial Statements” on page F-1 of this Annual Report.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
At the end of the period covered by this Annual Report, under the supervision and with the participation of our management, including our President and Chief Executive Officer and our Chief Financial Officer, we performed an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures. Based upon that evaluation, our President and Chief Executive Officer and Chief Financial Officer concluded that, as of the end of the period covered by this Annual Report, our disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting
The Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting (as such term is defined in the Exchange Act Rule 13a-15(f)). The Company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. GAAP, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding the prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements.
75
Internal control over financial reporting is designed to provide reasonable assurance to the Company’s management and board of directors regarding the preparation of reliable financial statements for external purposes in accordance with U.S. GAAP. Because of the inherent limitations in any internal control, no matter how well designed, misstatements may occur and not be prevented or detected. Accordingly, even effective internal control over financial reporting can provide only reasonable assurance with respect to financial statement preparation. Further, the evaluation of the effectiveness of internal control over financial reporting was made as of a specific date, and continued effectiveness in future periods is subject to the risks that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies and procedures may decline.
In connection with the preparation and filing of this Annual Report, the Company’s management, including our President and Chief Executive Officer and our Chief Financial Officer, conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2022, based on the framework set forth in “Internal Control—Integrated Framework (2013)” issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Based on its evaluation, the Company’s management concluded that, as of December 31, 2022, the Company’s internal control over financial reporting is effective based on the specified criteria.
Ernst & Young has issued an audit report on the effectiveness of our internal control over financial reporting, which follows this report.
Changes in Internal Control over Financial Reporting
There have not been any changes in our internal control over financial reporting during the fourth quarter of 2022 that have materially affected or are reasonably likely to materially affect, our internal control over financial reporting.
76
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and the Board of Directors of Orthofix Medical Inc.
Opinion on Internal Control over Financial Reporting
We have audited Orthofix Medical Inc.’s internal control over financial reporting as of December 31, 2022, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Orthofix Medical Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2022, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2022, and 2021, the related consolidated statements of operations and comprehensive income (loss), changes in shareholders' equity and cash flows for each of the three years in the period ended December 31, 2022, and the related notes and our report dated March 6, 2023, expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Dallas, Texas
March 6, 2023
77
Item 9B. Other Information
On March 3, 2023, Company entered into transition agreements with each of Jon Serbousek and Douglas Rice. Mr. Serbousek currently serves as the Company’s Executive Chairman and, prior to the completion on January 5, 2023 of the Company’s merger with SeaSpine Holdings Corporation, served as the Company’s President and Chief Executive Officer. Mr. Rice is currently providing assistance with integration activities, after previously having served as the Company’s Chief Financial Officer prior to the merger.
Under the transition agreement with Mr. Serbousek, the parties have agreed that Mr. Serbousek will not stand for re-election as a director at the Company’s 2023 annual meeting of stockholders (currently anticipated to be held in June 2023), at which point his service as Executive Chairman and a board member will cease, and he will remain employed in a non-officer capacity until July 5, 2023. The agreement provides that Mr. Serbousek will continue to receive his current annual base salary, and a pro-rated cash bonus for the 2023 calendar year equal to 105% of his annual base salary, for the portion of the 2023 calendar year he is employed. The agreement memorializes that as a result of the merger, Mr. Serbousek possesses “CiC Period Good Reason” under the terms of the change in control and severance agreement between Mr. Serbousek and the Company, and that Mr. Serbousek’s termination of employment on July 5, 2023 will be treated as a termination by Mr. Serbousek for “CiC Period Good Reason” under such change in control and severance agreement. The agreement also provides that the period for Mr. Serbousek to exercise outstanding stock options will be extended from 24 months to 48 months following such termination.
Under the transition agreement with Mr. Rice, the parties have agreed that Mr. Rice will continue to provide transition services as an employee until June 30, 2023, at which time his employment with the Company will cease. The agreement provides that in lieu of receiving an annual base salary and annual cash incentive program bonus opportunity for the 2023 calendar year, Mr. Rice will be paid a monthly fee of $65,000 through June 30, 2023. The agreement memorializes that as a result of the merger, Mr. Rice possesses “CiC Period Good Reason” under the terms of the change in control and severance agreement between Mr. Rice and the Company, and that if Mr. Rice serves through June 30, 2023, his termination of employment on such date will be treated as a termination by the Company without cause during a “CiC Period” under such change in control and severance agreement. The agreement also provides that the period for Mr. Rice to exercise outstanding stock options will be extended from 24 months to 48 months following such termination.
The foregoing descriptions of the transition agreements with Mr. Serbousek and Mr. Rice do not purport to be complete and are qualified in their entirety by reference to the full text of such agreements, which are filed respectively as Exhibits 10.70 and 10.71 hereto and incorporated herein by reference.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
None.
78
PART III
Information required by Items 10, 11, 12, 13 and 14 of Form 10-K is omitted from this Annual Report and will be filed in a definitive proxy statement or by an amendment to this Annual Report not later than 120 days after the end of the fiscal year covered by this Annual Report.
Item 10. Directors, Executive Officers and Corporate Governance
We will provide information that is responsive to this Item 10 regarding executive compensation in our definitive proxy statement or in an amendment to this Annual Report not later than 120 days after the end of the fiscal year covered by this Annual Report, in either case under the caption “Information About Directors,” “Section 16 (a) Beneficial Ownership Reporting Compliance” and others possibly elsewhere therein. That information is incorporated in this Item 10 by reference.
Item 11. Executive Compensation
We will provide information that is responsive to this Item 11 regarding executive compensation in our definitive proxy statement or in an amendment to this Annual Report not later than 120 days after the end of the fiscal year covered by this Annual Report, in either case under the caption “Executive Compensation,” and possibly elsewhere therein. That information is incorporated in this Item 11 by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
We will provide information that is responsive to this Item 12 regarding ownership of our securities by certain beneficial owners and our directors and executive officers, as well as information with respect to our equity compensation plans, in our definitive proxy statement or in an amendment to this Annual Report not later than 120 days after the end of the fiscal year covered by this Annual Report, in either case under the captions “Security Ownership of Certain Beneficial Owners and Management and Related Stockholders” and “Equity Compensation Plan Information,” and possibly elsewhere therein. That information is incorporated in this Item 12 by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
We will provide information that is responsive to this Item 13 regarding transactions with related parties and director independence in our definitive proxy statement or in an amendment to this Annual Report not later than 120 days after the end of the fiscal year covered by this Annual Report, in either case under the caption “Certain Relationships and Related Transactions,” and “Director Independence” and possibly elsewhere therein. That information is incorporated in this Item 13 by reference.
Item 14. Principal Accountant Fees and Services
We will provide information that is responsive to this Item 14 regarding principal accountant fees and services in our definitive proxy statement or in an amendment to this Annual Report not later than 120 days after the end of the fiscal year covered by this Annual Report, in either case under the caption “Principal Accountant Fees and Services,” and possibly elsewhere therein. That information is incorporated in this Item 14 by reference.
79
PART IV
Item 15. Exhibits, Financial Statement Schedules
(a) Documents filed as part of report on Form 10-K
The following documents are filed as part of this Annual Report on Form 10-K:
See “Index to Consolidated Financial Statements” on page F-1 of this Form 10-K.
2.Financial Statement Schedules
No schedules are required because either the required information is not present or is not present in amounts sufficient to require submission of the schedule, or because the information required is included in the consolidated financial statements or the notes thereto.
| | |
Exhibit
Number | | Description |
| | |
2.1 | | Agreement and Plan of Merger, dated as of October 10, 2022, by and among Orthofix Medical Inc., Orca Merger Sub Inc. and SeaSpine Holdings Corporation (filed as an Exhibit to the Company's Current Report on Form 8-K dated October 11, 2022 and incorporated herein by reference). |
| | |
2.2 | | Agreement and Plan of Merger, entered into March 15, 2018, by and among Blackstone Medical, Inc., Summit Development, Inc., and Spinal Kinetics, Inc. (filed as an exhibit to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2018 and incorporated herein by reference). |
| | |
3.1 | | Orthofix Medical Inc. Certificate of Incorporation (filed as an exhibit to the Company’s Current Report on Form 8-K dated August 1, 2018 and incorporated herein by reference). |
| | |
3.2 | | Amended and Restated Bylaws of Orthofix Medical Inc., as amended and restated effective October 10, 2022 (filed as an exhibit to the Company's Current Report on Form 8-K dated October 11, 2022 and incorporated herein by reference). |
| | |
4.1 | | Form of Stock Certificate (filed as an exhibit to the Company’s Current Report on Form 8-K dated August 1, 2018 and incorporated herein by reference). |
| | |
4.2 | | Description of the Registrant’s Securities Registered Pursuant to Section 12 of the Securities Exchange Act of 1934 (filed as an exhibit to the Company’s Annual Report on Form 10-K for the year ended December 31, 2019 and incorporated herein by reference). |
| | |
10.1 | | Second Amended and Restated Credit Agreement, dated October 25, 2019, among Orthofix Medical Inc., Orthofix Inc., Orthofix Spinal Implants Inc., Orthofix International B.V., Orthofix III B.V., and certain subsidiaries of Orthofix Medical Inc. as guarantors, the several banks and other financial institutions as may from time to time become parties thereunder as lenders, and JPMorgan Chase, N.A., as administrative agent (filed as an exhibit to the Company’s Current Report on Form 8-K filed on November 1, 2019 and incorporated herein by reference). |
| | |
10.2* | | First Amendment to Second Amended and Restated Credit Agreement, dated March 1, 2023, among Orthofix Medical Inc., Orthofix US LLC, Orthofix Netherlands B.V., and certain subsidiaries of Orthofix Medical Inc. as guarantors, the several banks and other financial institutions as may from time to time become parties thereunder as lenders, and JPMorgan Chase, N.A., as administrative agent. |
| | |
10.3† | | Amended and Restated Matrix Commercialization Collaboration Agreement, entered into as of February 7, 2022, by and between Orthofix US LLC and Muscoloskeletal Transplant Foundation Inc. (filed as an exhibit to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2022 and incorporated herein by reference. |
| | |
10.4† | | Supply Agreement between SeaSpine Orthopedics Corporation and PcoMed, LLC, dated March 1, 2021 (filed as an exhibit to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2021 by SeaSpine Holdings Corporation and incorporated herein by reference).
|
| | |
80
| | |
10.5* | | Lease Agreement between AR Industrial No. 1 Ltd. and Orthofix Inc. dated February 10, 2009. |
| | |
10.6* | | First Amendment to the Lease Agreement between AR Industrial No. 1 Ltd. and Orthofix Inc. dated April 13, 2009. |
| | |
10.7* | | Second Amendment to the Lease Agreement between AR Industrial No. 1 Ltd. and Orthofix Inc. dated May 12, 2010. |
| | |
10.8* | | Third Amendment to the Lease Agreement between AR Industrial No. 1 Ltd. and Orthofix Inc. dated December 21, 2017. |
| | |
10.9* | | Fourth Amendment to the Lease Agreement between AR Industrial No. 1 Ltd. and Orthofix Inc. dated March 13, 2018. |
| | |
10.10* | | Fifth Amendment to the Lease Agreement between AR Industrial No. 1 Ltd. and Orthofix Inc. dated January 3, 2019. |
| | |
10.11* | | Standard Lease Agreement between Lake Midas LLC and Spinal Kinetics, Inc. dated April 16, 2015. |
| | |
10.12* | | First Amendment to the Standard Lease Agreement between Lake Midas LLC and Spinal Kinetics LLC (formerly known as Spinal Kinetics, Inc.) dated March 4, 2022. |
| | |
10.13 | | Sublease Agreement between SeaSpine Orthopedics Corporation and SkinMedica, Inc., dated July 8, 2015 (filed as an exhibit to the Current Report on Form 8-K dated September 8, 2015 by SeaSpine Holdings Corporation and incorporated herein by reference).
|
| | |
10.14 | | Standard Industrial/Commercial Single-Tenant Lease–NET between Monarch RRC Properties, LP and Isotis Orthobiologics, Inc., dated June 1, 2022 (filed as an exhibit to the Quarterly Report on Form 10-Q for the quarter ended June 30, 2022 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.15 | | Orthofix Medical Inc. Second Amended and Restated Stock Purchase Plan, as amended by Amendment No. 1 thereto (filed as an exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2020 and incorporated herein by reference). |
| | |
10.16 | | Amendment No. 2 to the Orthofix Medical Inc. Second Amended and Restated Stock Purchase Plan (filed as an exhibit to the Company's Current Report on Form 8-K filed June 21, 2021 and incorporated by reference). |
| | |
10.17 | | Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (filed as an exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2018 and incorporated herein by reference). |
| | |
10.18 | | Amendment No. 1 to Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (filed as an exhibit to the Company’s Current Report on Form 8-K dated June 8, 2020 and incorporated herein by reference). |
| | |
10.19 | | Amendment No. 2 to Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (filed as an exhibit to the Company's Current Report on Form 8-K filed June 21, 2021 and incorporated by reference). |
| | |
10.20 | | Amendment No. 3 to the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (filed as an exhibit to the Company's Current Report on Form 8-K filed June 7, 2022 and incorporated by reference). |
| | |
10.21 | | Form of Employee Performance Stock Unit Agreement (2022 grant) under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (filed as an exhibit to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2022 and incorporated herein by reference). |
| | |
10.22 | | Form of Employee Performance Stock Unit Agreement (2016 – 2021 grants) under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (filed as an exhibit to the Company’s Annual Report on Form 10-K for the year ended December 31, 2019 and incorporated herein by reference). |
| | |
10.23* | | Form of Time-Based Vesting Employee Restricted Stock Unit Grant Agreement (2023 grant) under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan. |
| | |
10.24 | | Form of Time-Based Vesting Employee Restricted Stock Unit Grant Agreement (2018 – 2022 grants) under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (filed as an exhibit to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2022 and incorporated herein by reference). |
| | |
10.25* | | Form of Time-Based Vesting Employee Non-Qualified Stock Option Agreement (2023 grant) under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan. |
| | |
81
| | |
10.26 | | Form of Time-Based Vesting Employee Non-Qualified Stock Option Agreement under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (filed as an exhibit to the Company’s Current Report on Form 8-K filed July 8, 2016 and incorporated here by reference). |
| | |
10.27 | | Form of Employee Non-Qualified Stock Option Agreement under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan – July 2014-June 2016 (Time-Based Vesting) (filed as an exhibit to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2014 and incorporated herein by reference). |
| | |
10.28 | | Form of Employee Non-Qualified Stock Option Agreement under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (pre-2014 grants) (filed as an exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2012 and incorporated herein by reference). |
| | |
10.29* | | Form on Non-Employee Director Restricted Stock Unit Agreement (2023 grant) under the Orthofix Medical Inc. Amended and Restated 2023 Long-Term Incentive Plan. |
| | |
10.30 | | Form of Non-Employee Director Restricted Stock Unit Agreement (2017 - 2022 grants) under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (filed as an exhibit to the Company’s Form 10-Q filed on August 7, 2017 and incorporated herein by reference). |
| | |
10.31 | | Form of Time-Based Vesting Non-Employee Director Non-Qualified Stock Option Agreement under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (initial grant) (filed as an exhibit to the Company’s Current Report on Form 8-K filed July 8, 2016 and incorporated here by reference). |
| | |
10.32 | | Form of Non-Employee Director Non-Qualified Stock Option Agreement under the Orthofix Medical Inc. Amended and Restated 2012 Long-Term Incentive Plan (filed as an exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2012 and incorporated herein by reference). |
| | |
10.33 | | Employee Inducement Restricted Stock Unit Agreement for Jon Serbousek (filed as an exhibit to the Company’s Form S-8 filed on August 5, 2019 and incorporated herein by reference). |
| | |
10.34 | | Employee Inducement Non-Qualified Stock Option Agreement for Jon Serbousek (filed as an exhibit to the Company’s Form S-8 filed on August 5, 2019 and incorporated herein by reference). |
| | |
10.35 | | Inducement Grant Non-Qualified Stock Option Agreement, dated March 13, 2013, between Orthofix International N.V. and Bradley R. Mason (filed as an exhibit to the Company’s Current Report on Form 8-K filed March 13, 2013 and incorporated herein by reference). |
| | |
10.36 | | Orthofix Medical Inc. Inducement Plan for SeaSpine Employees (filed as Exhibit 4.3 to the Company's Registration Statement on Form S-8 (Registration No. 333-269116) filed January 4, 2023 and incorporated herein by reference). |
| | |
10.37 | | Orthofix Medical Inc. Inducement Plan for SeaSpine Employees – Stock Unit Grant Agreement (filed as Exhibit 4.4 to the Company's Registration Statement on Form S-8 (Registration No. 333-269116) filed January 4, 2023 and incorporated herein by reference). |
| | |
10.38 | | Orthofix Medical Inc. Inducement Plan for SeaSpine Employees – Nonqualified Stock Option Grant Agreement (filed as Exhibit 4.5 to the Company's Registration Statement on Form S-8 (Registration No. 333-269116) filed January 4, 2023 and incorporated herein by reference). |
| | |
10.39 | | SeaSpine Holdings Corporation Amended and Restated 2015 Incentive Award Plan (As Amended and Restated as of March 30, 2016) (filed as an exhibit to the Company’s Form S-8 filed on January 10, 2023 and incorporated herein by reference). |
| | |
10.40 | | First Amendment to the SeaSpine Holdings Corporation Amended and Restated 2015 Incentive Award Plan (filed as an exhibit to the Company’s Form S-8 filed on January 10, 2023 and incorporated herein by reference). |
| | |
10.41 | | Second Amendment to the SeaSpine Holdings Corporation Amended and Restated 2015 Incentive Award Plan (filed as an exhibit to the Company’s Form S-8 filed on January 10, 2023 and incorporated herein by reference). |
| | |
10.42 | | Amendment to the SeaSpine Holdings Corporation Amended and Restated 2015 Incentive Award Plan (filed as an exhibit to the Company’s Form S-8 filed on January 10, 2023 and incorporated herein by reference). |
| | |
82
| | |
10.43 | | Form of Stock Option Grant Notice and Stock Option Agreement under SeaSpine Holdings Corporation 2015 Incentive Award Plan (three-month exercise period post-termination) (filed as an exhibit to the Registration Statement on Form S-8 filed with the Commission on June 7, 2016 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.44 | | Form of Stock Option Grant Notice and Stock Option Agreement under SeaSpine Holdings Corporation 2015 Incentive Award Plan (one-year exercise period post-termination) (filed as an exhibit to Amendment No. 2 to Form 10 filed with the Commission on June 1, 2015 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.45 | | Form of Restricted Stock Award Grant Notice and Restricted Stock Award Agreement under SeaSpine Holdings Corporation 2015 Incentive Award Plan (filed as an exhibit to the Registration Statement on Form S-8 filed with the Commission on June 7, 2016 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.46 | | Form of Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement under SeaSpine Holdings Corporation 2015 Incentive Award Plan (filed as an exhibit to the Annual Report on Form 10-K for the year ended December 31, 2016 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.47 | | Form of Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement under SeaSpine Holdings Corporation 2015 Incentive Award Plan (grants awarded after February 1, 2018) (filed as an exhibit to the Annual Report on Form 10-K for the year ended December 31, 2017 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.48 | | Form of Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement under SeaSpine Holdings Corporation 2015 Incentive Award Plan (grants awarded after January 1, 2020) (filed as an exhibit to the Annual Report on Form 10-K for the year ended December 31, 2019 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.49 | | Form of Stock Option Grant Notice and Stock Option Agreement under SeaSpine Holdings Corporation 2015 Incentive Award Plan (grants to Senior Leadership Team Members awarded after June 6, 2018) (filed as an exhibit to the Quarterly Report on Form 10-Q for the quarter ended June 30, 2018 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.50 | | Form of Stock Option Grant Notice and Stock Option Agreement under SeaSpine Holdings Corporation 2015 Incentive Award Plan (grants to Non-Senior Leadership Team Members awarded after June 6, 2018) (filed as an exhibit to the Quarterly Report on Form 10-Q for the quarter ended June 30, 2018 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.51 | | Annual Incentive Program under SeaSpine Holdings Corporation Amended and Restated 2015 Incentive Award Plan, dated January 1, 2019 (filed as an exhibit to the Current Report on Form 8-K dated January 28, 2021 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.52 | | SeaSpine Holdings Corporation 2018 Employment Inducement Incentive Award Plan (filed as an exhibit to the Company’s Form S-8 filed on January 10, 2023 and incorporated herein by reference). |
| | |
10.53 | | Form of Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement under SeaSpine Holdings Corporation 2018 Employment Inducement Incentive Award Plan (filed as an exhibit to the Quarterly Report on Form 10-Q for the quarter ended June 30, 2018 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.54 | | Form of Stock Option Grant Notice and Stock Option Agreement under SeaSpine Holdings Corporation 2018 Employment Inducement Incentive Award Plan (grants to Senior Leadership Team Members) (filed as an exhibit to the Quarterly Report on Form 10-Q for the quarter ended June 30, 2018 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.55 | | Form of Stock Option Grant Notice and Stock Option Agreement under SeaSpine Holdings Corporation 2018 Employment Inducement Incentive Award Plan (grants to Non-Senior Leadership Team Members) (filed as an exhibit to the Quarterly Report on Form 10-Q for the quarter ended June 30, 2018 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.56 | | SeaSpine Holdings Corporation 2020 Employment Inducement Incentive Award Plan (filed as an exhibit to the Company’s Form S-8 filed on January 10, 2023 and incorporated herein by reference). |
| | |
83
| | |
10.57 | | Form of Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement under SeaSpine Holdings Corporation 2020 Employment Inducement Incentive Award Plan (filed as an exhibit to the Quarterly Report on Form 10-Q for the quarter ended June 30, 2020 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.58 | | Form of Stock Option Grant Notice and Stock Option Agreement under SeaSpine Holdings Corporation 2020 Employment Inducement Incentive Award Plan (grants to Senior Leadership Team Members) (filed as an exhibit to the Quarterly Report on Form 10-Q for the quarter ended June 30, 2020 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.59 | | Form of Stock Option Grant Notice and Stock Option Agreement under SeaSpine Holdings Corporation 2020 Employment Inducement Incentive Award Plan (grants to Non-Senior Leadership Team Members) (filed as an exhibit to the Quarterly Report on Form 10-Q for the quarter ended June 30, 2020 by SeaSpine Holdings Corporation and incorporated herein by reference). |
| | |
10.60 | | Form of Indemnification Agreement between Orthofix Medical Inc. and its directors and officers (filed as an exhibit to the Company's Current Report on Form 8-K filed January 5, 2023 and incorporated herein by reference). |
| | |
10.61 | | Form of Indemnification Agreement between Orthofix Medical Inc. and its directors and officers (incorporated by reference to Exhibit 10.1 to the Company’s Registration Statement on Form S-4 (Registration No. 333-224407) filed April 23, 2018). |
| | |
10.62 | | Transition and Retirement Agreement, dated February 25, 2019, between Bradley R. Mason and Orthofix Medical Inc. (filed as an exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2018 and incorporated herein by reference). |
| | |
10.63 | | Change in Control and Severance Agreement, dated November 1, 2019, between Orthofix Medical Inc. and Jon Serbousek (filed as an Exhibit to the Company’s Current Report on Form 8-K filed November 1, 2019 and incorporated herein by reference). |
| | |
10.64 | | Change in Control and Severance Agreement, dated November 1, 2019, between Orthofix Medical Inc. and Kevin Kenny (filed as an exhibit to the Company’s Annual Report on Form 10-K for the year ended December 31, 2019 and incorporated herein by reference). |
| | |
10.65 | | Amended Change in Control and Severance Agreement, dated November 1, 2016, between Orthofix International N.V. and Doug Rice (filed as an exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2016 and incorporated herein by reference). |
| | |
10.66 | | Change in Control and Severance Agreement, dated November 1, 2016, between Orthofix International N.V. and Kimberley Elting (filed as an exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2016 and incorporated herein by reference). |
| | |
10.67 | | Offer Letter between the Company and Keith C. Valentine (filed as an exhibit to the Company's Current Report on Form 8-K filed on January 5, 2023 and incorporated herein by reference). |
| | |
10.68 | | Offer Letter between the Company and John J. Bostjancic (filed as an exhibit to the Company's Current Report on Form 8-K filed on January 5, 2023 and incorporated herein by reference). |
| | |
10.69 | | Offer Letter between the Company and Patrick L. Keran (filed as an exhibit to the Company's Current Report on Form 8-K filed on January 5, 2023 and incorporated herein by reference). |
| | |
10.70* | | Transition Agreement, dated March 3, 2023, between Orthofix Medical Inc. and Jon Serbousek. |
| | |
10.71* | | Transition Agreement, dated March 3, 2023, between Orthofix Medical Inc. and Doug Rice. |
| | |
21.1* | | List of Subsidiaries. |
| | |
23.1* | | Consent of Independent Registered Public Accounting Firm. |
| | |
31.1* | | Rule 13a-14(a)/15d-14(a) Certification of Chief Executive Officer. |
| | |
31.2* | | Rule 13a-14(a)/15d-14(a) Certification of Chief Financial Officer. |
| | |
32.1* | | Section 1350 Certification of Chief Executive Officer and Certification of Chief Financial Officer. |
84
| | |
| | |
101.INS | | Inline XBRL Instance Document – the instance document does not appear in the interactive Data File because its XBRL tags are embedded within the XBRL document. |
| | |
101.SCH* | | Inline XBRL Taxonomy Extension Schema Document. |
| | |
101.CAL* | | Inline XBRL Taxonomy Calculation Linkbase Document. |
| | |
101.DEF* | | Inline XBRL Taxonomy Definition Linkbase Document. |
| | |
101.LAB* | | Inline XBRL Taxonomy Label Linkbase Document. |
| | |
101.PRE* | | Inline XBRL Taxonomy Presentation Linkbase Document. |
| | |
104 | | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101). |
* Filed with this Form 10-K.
† Certain private or confidential portions of this exhibit that are not material were omitted by means of redacting a portion of the text and replacing it with a bracketed asterisk.
Item 16. Form 10-K Summary
None
85
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | |
| | | | ORTHOFIX MEDICAL INC. |
| | | | |
Dated: March 6, 2023 | | | | By: | | /s/ KEITH VALENTINE |
| | | | Name: | | Keith Valentine |
| | | | Title: | | President and Chief Executive Officer, Director |
| | | | | | |
Dated: March 6, 2023 | | | | By: | | /s/ JOHN BOSTJANCIC |
| | | | Name: | | John Bostjancic |
| | | | Title: | | Chief Financial Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| | | | |
Name | | Title | | Date |
| | | | |
/s/ KEITH VALENTINE Keith Valentine | | President and Chief Executive Officer, Director (Principal Executive Officer) | | March 6, 2023 |
| | | | |
/s/ JOHN BOSTJANCIC John Bostjancic | | Chief Financial Officer (Principal Financial and Accounting Officer) | | March 6, 2023 |
| | | | |
/s/ CATHERINE BURZIK Catherine Burzik | | Lead Independent Director of the Board | | March 6, 2023 |
| | | | |
/s/ JON SERBOUSEK Jon Serbousek | | Director, Executive Chairman of the Board | | March 6, 2023 |
| | | | |
/s/ STUART ESSIG Stuart Essig | | Director | | March 6, 2023 |
| | | | |
/s/ JASON HANNON Jason Hannon | | Director | | March 6, 2023 |
| | | | |
/s/ JOHN HENNEMAN, III John Henneman, III | | Director | | March 6, 2023 |
| | | | |
/s/ JAMES HINRICHS | | Director | | March 6, 2023 |
James Hinrichs |
| | | | |
/s/ SHWETA SINGH MANIAR | | Director | | March 6, 2023 |
Shweta Singh Maniar |
| | | | |
/s/ MICHAEL PAOLUCCI | | Director | | March 6, 2023 |
Michael Paolucci | | | | |
| | | | |
86
ORTHOFIX MEDICAL INC.
Statement of Management’s Responsibility for Financial Statements
To the Shareholders of Orthofix Medical Inc.:
Management is responsible for the preparation of the consolidated financial statements and related information that are presented in this Annual Report. The consolidated financial statements, which include amounts based on management’s estimates and judgments, have been prepared in conformity with accounting principles generally accepted in the United States. Other financial information in the report to shareholders is consistent with that in the consolidated financial statements.
The Company maintains accounting and internal control systems to provide reasonable assurance at a reasonable cost that assets are safeguarded against loss from unauthorized use or disposition, and that the financial records are reliable for preparing financial statements and maintaining accountability for assets. These systems are augmented by written policies, an organizational structure providing division of responsibilities, and careful selection and training of qualified personnel.
The Company engaged Ernst & Young LLP, independent registered public accountants, to audit and render an opinion on the consolidated financial statements in accordance with auditing standards of the Public Company Accounting Oversight Board (United States). These standards include an assessment of the systems of internal controls and tests of transactions to the extent considered necessary by them to support their opinion.
The Board of Directors, through its Audit Committee, consisting solely of outside directors of the Company, meets periodically with management and our independent registered public accountants to ensure that each is meeting its responsibilities and to discuss matters concerning internal controls and financial reporting. Ernst & Young LLP has full and free access to the Audit Committee.
James Hinrichs
Chairman of the Audit Committee
Keith Valentine
President and Chief Executive Officer, Director
John Bostjancic
Chief Financial Officer
87
ORTHOFIX MEDICAL INC.
Index to Consolidated Financial Statements
| | | |
| Page | |
Index to Consolidated Financial Statements | | F-1 | |
Report of Independent Registered Public Accounting Firm (PCAOB ID: 42) | | F-2 | |
Consolidated Balance Sheets as of December 31, 2022 and 2021 | | F-4 | |
Consolidated Statements of Operations and Comprehensive Income (Loss) for the years ended December 31, 2022, 2021, and 2020 | | F-5 | |
Consolidated Statements of Changes in Shareholders’ Equity for the years ended December 31, 2022, 2021, and 2020 | | F-6 | |
Consolidated Statements of Cash Flows for the years ended December 31, 2022, 2021, and 2020 | | F-7 | |
Notes to the Consolidated Financial Statements | | F-8 | |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and the Board of Directors of Orthofix Medical Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Orthofix Medical Inc. (the Company) as of December 31, 2022 and 2021, the related consolidated statements of operations and comprehensive income (loss), changes in shareholders' equity and cash flows for each of the three years in the period ended December 31, 2022, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2022 and 2021, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2022, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2022, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 6, 2023, expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the account or disclosure to which it relates.
F-2
| |
Inventory Excess and Obsolescence Reserves |
Description of the Matter | At December 31, 2022, the Company’s inventory balance is $100.2 million, which is net of management’s estimate of inventory excess and obsolescence reserves. As described in Note 5 to the consolidated financial statements, management adjusts the value of its inventory to net realizable value to the extent it determines inventory cost cannot be recovered due to obsolescence or other factors. In order to make these determinations, management estimates future demand to determine the appropriate inventory reserves and to make corresponding adjustments to the carrying value of these inventories to reflect the lower of cost or net realizable value. Auditing management’s estimate of the inventory excess and obsolescence reserves involved a high degree of subjectivity because the estimate was sensitive to changes in assumptions, including estimated product demand, length of product life cycles, and the period required to evaluate the level of market acceptance for new products. These assumptions have a significant effect on the measurement of inventory excess and obsolescence reserves. |
How We Addressed the Matter in Our Audit | We obtained an understanding, evaluated the design and tested the operating effectiveness of controls that address the risks of material misstatement relating to the measurement and valuation of inventory excess and obsolescence reserves. For example, we tested controls over the Company’s processes to estimate the inventory excess and obsolescence reserves, management’s review and approval of the model used to estimate the inventory excess and obsolescence reserve, including the data inputs and outputs of such model and management’s qualitative adjustments to the model. To test the inventory excess and obsolescence reserve balance, we performed audit procedures that included, among others, evaluating the significant assumptions and qualitative adjustments described above and the underlying data used by the Company in its analysis. Our audit procedures included testing the completeness and accuracy of the underlying data used in the model and evaluating whether such data was representative of current circumstances. We assessed the historical accuracy of management’s estimates and performed sensitivity analyses of significant assumptions to evaluate the changes in the inventory excess and obsolescence reserves that would result from changes in the assumptions. |
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2002.
Dallas, Texas
March 6, 2023
F-3
ORTHOFIX MEDICAL INC.
Consolidated Balance Sheets as of December 31, 2022 and 2021
| | | | | | | | |
(U.S. Dollars, in thousands, except par value data) | | 2022 | | | 2021 | |
Assets | | | | | | |
Current assets | | | | | | |
Cash and cash equivalents | | $ | 50,700 | | | $ | 87,847 | |
Accounts receivable, net of allowances of $6,419 and $4,944, respectively | | | 82,857 | | | | 78,560 | |
Inventories | | | 100,150 | | | | 82,974 | |
Prepaid expenses and other current assets | | | 22,283 | | | | 20,141 | |
Total current assets | | | 255,990 | | | | 269,522 | |
Property, plant and equipment, net | | | 58,229 | | | | 59,252 | |
Intangible assets, net | | | 47,388 | | | | 52,666 | |
Goodwill | | | 71,317 | | | | 71,317 | |
Other long-term assets | | | 25,705 | | | | 23,866 | |
Total assets | | $ | 458,629 | | | $ | 476,623 | |
Liabilities and shareholders’ equity | | | | | | |
Current liabilities | | | | | | |
Accounts payable | | $ | 27,598 | | | $ | 26,459 | |
Current portion of finance lease liability | | | 652 | | | | 2,590 | |
Other current liabilities | | | 55,374 | | | | 76,781 | |
Total current liabilities | | | 83,624 | | | | 105,830 | |
Long-term portion of finance lease liability | | | 19,239 | | | | 19,890 | |
Other long-term liabilities | | | 18,906 | | | | 13,969 | |
Total liabilities | | | 121,769 | | | | 139,689 | |
Contingencies (Note 13) | | | | | | |
Shareholders’ equity | | | | | | |
Common shares $0.10 par value; 50,000 shares authorized;
20,162 and 19,837 issued and outstanding as of December 31,
2022 and 2021, respectively | | | 2,016 | | | | 1,983 | |
Additional paid-in capital | | | 334,969 | | | | 313,951 | |
Retained earnings | | | 1,251 | | | | 21,000 | |
Accumulated other comprehensive loss | | | (1,376 | ) | | | — | |
Total shareholders’ equity | | | 336,860 | | | | 336,934 | |
Total liabilities and shareholders’ equity | | $ | 458,629 | | | $ | 476,623 | |
The accompanying notes form an integral part of these consolidated financial statements.
F-4
ORTHOFIX MEDICAL INC.
Consolidated Statements of Operations and Comprehensive Income (Loss)
For the years ended December 31, 2022, 2021, and 2020
| | | | | | | | | | | | |
(U.S. Dollars, in thousands, except share and per share data) | | 2022 | | | 2021 | | | 2020 | |
Net sales | | $ | 460,713 | | | $ | 464,479 | | | $ | 406,562 | |
Cost of sales | | | 123,544 | | | | 114,914 | | | | 101,889 | |
Gross profit | | | 337,169 | | | | 349,565 | | | | 304,673 | |
Sales and marketing | | | 228,810 | | | | 221,318 | | | | 204,434 | |
General and administrative | | | 79,966 | | | | 69,353 | | | | 67,948 | |
Research and development | | | 49,065 | | | | 49,621 | | | | 39,056 | |
Acquisition-related amortization and remeasurement | | | (7,404 | ) | | | 17,588 | | | | (499 | ) |
Operating (loss) | | | (13,268 | ) | | | (8,315 | ) | | | (6,266 | ) |
Interest expense, net | | | (1,288 | ) | | | (1,837 | ) | | | (2,483 | ) |
Other income (expense), net | | | (3,150 | ) | | | (3,343 | ) | | | 8,381 | |
(Loss) before income taxes | | | (17,706 | ) | | | (13,495 | ) | | | (368 | ) |
Income tax benefit (expense) | | | (2,043 | ) | | | (24,884 | ) | | | 2,885 | |
Net income (loss) | | $ | (19,749 | ) | | $ | (38,379 | ) | | $ | 2,517 | |
| | | | | | | | | |
Net income (loss) per common share: | | | | | | | | | |
Basic | | $ | (0.98 | ) | | $ | (1.95 | ) | | $ | 0.13 | |
Diluted | | | (0.98 | ) | | | (1.95 | ) | | | 0.13 | |
Weighted average number of common shares: | | | | | | | | | |
Basic | | | 20,053,548 | | | | 19,690,593 | | | | 19,267,920 | |
Diluted | | | 20,053,548 | | | | 19,690,593 | | | | 19,391,718 | |
| | | | | | | | | |
Other comprehensive income (loss), before tax | | | | | | | | | |
Unrealized gain (loss) on debt securities | | | 395 | | | | (942 | ) | | | 1,881 | |
Currency translation adjustment | | | (1,771 | ) | | | (2,544 | ) | | | 4,872 | |
Other comprehensive income (loss), before tax | | | (1,376 | ) | | | (3,486 | ) | | | 6,753 | |
Income tax benefit (expense) related to items of other comprehensive income (loss) | | | — | | | | 234 | | | | (462 | ) |
Other comprehensive income (loss), net of tax | | | (1,376 | ) | | | (3,252 | ) | | | 6,291 | |
Comprehensive income (loss) | | $ | (21,125 | ) | | $ | (41,631 | ) | | $ | 8,808 | |
The accompanying notes form an integral part of these consolidated financial statements.
F-5
ORTHOFIX MEDICAL INC.
Consolidated Statements of Changes in Shareholders’ Equity
For the years ended December 31, 2022, 2021, and 2020
| | | | | | | | | | | | | | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | Number of
Common
Shares
Outstanding | | | Common
Shares | | | Additional
Paid-in
Capital | | | Retained
Earnings | | | Accumulated
Other
Comprehensive
Income (Loss) | | | Total
Shareholders’
Equity | |
At December 31, 2019 | | | 19,023 | | | $ | 1,902 | | | $ | 271,019 | | | $ | 57,749 | | | $ | (3,039 | ) | | $ | 327,631 | |
Cumulative effect adjustment from adoption
of ASU 2016-13 | | | — | | | | — | | | | — | | | | (887 | ) | | | — | | | | (887 | ) |
Net income | | | — | | | | — | | | | — | | | | 2,517 | | | | — | | | | 2,517 | |
Other comprehensive income, net of tax | | | — | | | | — | | | | — | | | | — | | | | 6,291 | | | | 6,291 | |
Share-based compensation expense | | | — | | | | — | | | | 16,207 | | | | — | | | | — | | | | 16,207 | |
Common shares issued, net | | | 401 | | | | 40 | | | | 5,065 | | | | — | | | | — | | | | 5,105 | |
At December 31, 2020 | | | 19,424 | | | $ | 1,942 | | | $ | 292,291 | | | $ | 59,379 | | | $ | 3,252 | | | $ | 356,864 | |
Net loss | | | — | | | | — | | | | — | | | | (38,379 | ) | | | — | | | | (38,379 | ) |
Other comprehensive loss, net of tax | | | — | | | | — | | | | — | | | | — | | | | (3,252 | ) | | | (3,252 | ) |
Share-based compensation expense | | | — | | | | — | | | | 15,432 | | | | — | | | | — | | | | 15,432 | |
Common shares issued, net | | | 413 | | | | 41 | | | | 6,228 | | | | — | | | | — | | | | 6,269 | |
At December 31, 2021 | | | 19,837 | | | $ | 1,983 | | | $ | 313,951 | | | $ | 21,000 | | | $ | - | | | $ | 336,934 | |
Net loss | | | — | | | | — | | | | — | | | | (19,749 | ) | | | — | | | | (19,749 | ) |
Other comprehensive loss, net of tax | | | — | | | | — | | | | — | | | | — | | | | (1,376 | ) | | | (1,376 | ) |
Share-based compensation expense | | | — | | | | — | | | | 18,443 | | | | — | | | | — | | | | 18,443 | |
Common shares issued, net | | | 325 | | | | 33 | | | | 2,575 | | | | — | | | | — | | | | 2,608 | |
At December 31, 2022 | | | 20,162 | | | $ | 2,016 | | | $ | 334,969 | | | $ | 1,251 | | | $ | (1,376 | ) | | $ | 336,860 | |
The accompanying notes form an integral part of these consolidated financial statements.
F-6
ORTHOFIX MEDICAL INC.
Consolidated Statements of Cash Flows
For the years ended December 31, 2022, 2021, and 2020
| | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Cash flows from operating activities | | | | | | | | | |
Net income (loss) | | $ | (19,749 | ) | | $ | (38,379 | ) | | $ | 2,517 | |
Adjustments to reconcile net income (loss) to net cash from operating activities | |
Depreciation and amortization | | | 29,019 | | | | 29,599 | | | | 30,546 | |
Impairment of goodwill | | | — | | | | 11,756 | | | | — | |
Amortization of operating lease assets, debt costs, and other assets | | | 3,056 | | | | 3,496 | | | | 3,730 | |
Provision for expected credit losses | | | 2,095 | | | | 444 | | | | 199 | |
Deferred income taxes | | | 314 | | | | 24,482 | | | | 10,787 | |
Share-based compensation expense | | | 18,443 | | | | 15,432 | | | | 16,207 | |
Interest and (gain) loss on the valuation of investment securities | | | (308 | ) | | | (1,146 | ) | | | 116 | |
Change in fair value of contingent consideration | | | (17,200 | ) | | | (3,575 | ) | | | (7,300 | ) |
Other | | | 2,027 | | | | 1,064 | | | | (2,228 | ) |
Changes in operating assets and liabilities, net of effects of acquisitions | | | | | | | | | |
Accounts receivable | | | (6,735 | ) | | | (7,049 | ) | | | 13,283 | |
Inventories | | | (18,133 | ) | | | 619 | | | | (873 | ) |
Prepaid expenses and other current assets | | | (874 | ) | | | (2,834 | ) | | | 4,526 | |
Accounts payable | | | 2,282 | | | | 4,253 | | | | 2,532 | |
Other current liabilities | | | 627 | | | | 1,013 | | | | 5,975 | |
Contract liability (Note 15) | | | (4,791 | ) | | | (9,060 | ) | | | 13,851 | |
Payment of contingent consideration | | | — | | | | (6,595 | ) | | | — | |
Other long-term assets and liabilities | | | (1,611 | ) | | | (5,045 | ) | | | (19,596 | ) |
Net cash from operating activities | | | (11,538 | ) | | | 18,475 | | | | 74,272 | |
Cash flows from investing activities | | | | | | | | | |
Acquisition of a business | | | — | | | | — | | | | (18,000 | ) |
Capital expenditures for property, plant and equipment | | | (21,364 | ) | | | (17,785 | ) | | | (15,485 | ) |
Capital expenditures for intangible assets | | | (1,796 | ) | | | (1,807 | ) | | | (1,609 | ) |
Purchase of investment securities | | | — | | | | (2,171 | ) | | | (10,000 | ) |
Asset acquisitions and other investments | | | (1,374 | ) | | | (1,250 | ) | | | (7,240 | ) |
Net cash from investing activities | | | (24,534 | ) | | | (23,013 | ) | | | (52,334 | ) |
Cash flows from financing activities | | | | | | | | | |
Proceeds from revolving credit facility | | | — | | | | — | | | | 100,000 | |
Repayment of revolving credit facility | | | — | | | | — | | | | (100,000 | ) |
Proceeds from issuance of common shares | | | 4,337 | | | | 8,824 | | | | 7,598 | |
Payments related to withholdings for share-based compensation | | | (1,729 | ) | | | (2,555 | ) | | | (2,493 | ) |
Payment of contingent consideration | | | — | | | | (8,405 | ) | | | — | |
Payments related to finance lease obligation | | | (2,594 | ) | | | (537 | ) | | | (323 | ) |
Payment of debt issuance costs and other financing activities | | | (92 | ) | | | (948 | ) | | | (1,537 | ) |
Net cash from financing activities | | | (78 | ) | | | (3,621 | ) | | | 3,245 | |
Effect of exchange rate changes on cash and restricted cash | | | (997 | ) | | | (815 | ) | | | 1,235 | |
Net change in cash, cash equivalents, and restricted cash | | | (37,147 | ) | | | (8,974 | ) | | | 26,418 | |
Cash, cash equivalents, and restricted cash at the beginning of the year | | | 87,847 | | | | 96,821 | | | | 70,403 | |
Cash, cash equivalents, and restricted cash at the end of the year | | $ | 50,700 | | | $ | 87,847 | | | $ | 96,821 | |
| | | | | | | | | |
Components of cash, cash equivalents, and restricted cash at the end of the year | |
Cash and cash equivalents | | $ | 50,700 | | | $ | 87,847 | | | $ | 96,291 | |
Restricted cash | | | — | | | | — | | | | 530 | |
Cash, cash equivalents, and restricted cash at the end of the year | | $ | 50,700 | | | $ | 87,847 | | | $ | 96,821 | |
The accompanying notes form an integral part of these consolidated financial statements
F-7
ORTHOFIX MEDICAL INC.
Notes to the Consolidated Financial Statements
1. Business and basis of presentation
Description of the Business
Orthofix Medical Inc. and its subsidiaries (the “Company”), following its recent merger with SeaSpine Holdings Corporation ("SeaSpine"), is a leading global spine and orthopedics company with a comprehensive portfolio of biologics, innovative spinal hardware, bone growth therapies, specialized orthopedic solutions and a leading surgical navigation system. Its products are distributed in 68 countries worldwide.
The Company is headquartered in Lewisville, Texas, and has primary offices in Carlsbad, CA, with a focus on spinal product innovation and surgeon education, and in Verona, Italy, with an emphasis on product innovation, production, and medical education for Orthopedics. The combined Company’s global R&D, commercial and manufacturing footprint also includes facilities and offices in Irvine, CA, Toronto, Canada, Sunnyvale, CA, Wayne, PA, Olive Branch, MS, Maidenhead, UK, Munich, Germany, Paris, France and Sao Paulo, Brazil.
The merger with SeaSpine was completed on January 5, 2023, with SeaSpine continuing as a wholly-owned subsidiary of Orthofix following the transaction. For additional discussion of the merger with SeaSpine, see Note 22. Orthofix, as the corporate parent entity in the combined company structure, will continue to trade on NASDAQ under the symbol “OFIX.” The combined company will be renamed at a later date and until then will continue to be known as Orthofix Medical Inc. The financial statements of the Company for the period ended as of December 31, 2022, do not include the financial position or operations of SeaSpine since the merger occurred subsequent to the end of the reporting period.
Basis of Presentation
The consolidated financial statements include the financial statements of the Company and its wholly owned subsidiaries. All intercompany accounts and transactions are eliminated in consolidation. Information on our accounting policies and methods used in the preparation of our consolidated financial statements are included, where applicable, in the respective footnotes that follow.
| | |
Footnote | | Footnote Reference |
Business and basis of presentation | | 1 |
Significant accounting policies | | 2 |
Recently adopted accounting standards, recently issued accounting pronouncements, and recent law changes | | 3 |
Acquisitions | | 4 |
Inventories | | 5 |
Property, plant, and equipment | | 6 |
Intangible assets | | 7 |
Goodwill | | 8 |
Leases | | 9 |
Other current liabilities | | 10 |
Long-term debt | | 11 |
Fair value measurements and investments | | 12 |
Commitments and contingencies | | 13 |
Shareholders' equity | | 14 |
Revenue recognition and accounts receivable | | 15 |
Business segment information | | 16 |
Acquisition-related amortization and remeasurement | | 17 |
Share-based compensation | | 18 |
Defined contribution plans and deferred compensation | | 19 |
Income taxes | | 20 |
Earnings per share | | 21 |
Subsequent events | | 22 |
F-8
2. Significant accounting policies
The preparation of financial statements in conformity with United States generally accepted accounting principles (“U.S. GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. On an ongoing basis, we evaluate these estimates, including those related to contractual allowances, allowances for expected credit losses, inventories, valuation of intangible assets, goodwill, fair value measurements, litigation and contingent liabilities, income taxes, and share-based compensation. We base our estimates on historical experience, future expectations, and other relevant assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates.
The following is a discussion of accounting policies and methods used in our consolidated financial statements that are not presented within other footnotes.
Market risk
In the ordinary course of business, the Company is exposed to the impact of changes in interest rates and foreign currency fluctuations. The Company’s objective is to limit the impact of such movements on earnings and cash flows. In order to achieve this objective, the Company seeks to balance its non-U.S. Dollar denominated income and expenditures.
The financial statements for operations outside the U.S. are generally maintained in their local currency. All foreign currency denominated balance sheet accounts, except shareholders’ equity, are translated to U.S. Dollars at year end exchange rates, and revenue and expense items are translated at average rates of exchange prevailing during the year. Gains and losses resulting from the translation of foreign currency are recorded in the accumulated other comprehensive income (loss) component of shareholders’ equity. Transactional foreign currency gains and losses, including those generated from intercompany operations, are included in other expense, net and were a loss of $3.3 million, a loss of $4.0 million, and a gain of $3.9 million for the years ended December 31, 2022, 2021, and 2020, respectively.
Financial instruments and concentration of credit risk
Financial instruments that could subject the Company to a concentration of credit risk consist primarily of cash, cash equivalents, and accounts receivable. Generally, cash is held at large financial institutions and cash equivalents consist of highly liquid money market funds. The Company performs ongoing credit evaluations of customers, generally does not require collateral, and maintains a reserve for expected credit losses. The Company believes that a concentration of credit risk related to accounts receivable is limited because customers are geographically dispersed and end users are diversified.
Cash, cash equivalents, and restricted cash
The Company considers all highly liquid investments with an original maturity of three months or less to be cash equivalents.
In September 2019, approximately $0.5 million (based upon foreign exchange rates as of December 31, 2020) of the Company’s cash in Brazil was frozen upon request to satisfy a judgment related to an ongoing legal dispute with a former Brazilian distributor. In December 2021, the dispute was settled and the cash was disbursed to the former distributor.
Investing activities that did not result in cash receipts or cash payments during the years ended December 31, 2022, 2021, and 2020 consisted of the following, which were not included within cash from investing activities in the Company’s consolidated statements of cash flows:
| | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Supplemental disclosure of cash flow information: | | | | | | | | | |
Noncash investing activities: | | | | | | | | | |
Intangible assets acquired in asset acquisitions | | $ | 2,000 | | | $ | — | | | $ | 1,575 | |
Contingent consideration recognized at acquisition date | | | — | | | | — | | | | 375 | |
F-9
Advertising costs
Advertising costs are expensed as incurred. Advertising costs are included within sales and marketing expense and totaled $0.5 million, $0.5 million, and $0.9 million for the years ended December 31, 2022, 2021, and 2020, respectively.
Research and development costs, including collaborative arrangements
Expenditures for research and development are expensed as incurred. Expenditures related to the Company’s collaborative arrangement with MTF Biologics (“MTF”) are expensed based on the terms of the related agreement. The Company recognized $0.0 million, $0.8 million and $0.8 million in research and development expense for the years ended December 31, 2022, 2021, and 2020, respectively.
In October 2020, the Company and Neo Medical SA, a privately held Swiss-based company developing a new generation of products for spinal surgery (“Neo Medical”), entered into a co-development agreement covering the parties’ joint development of single use instruments for cervical spine procedures. In connection with this agreement, the Company is responsible for the payment of variable costs associated with the development of the specified products. Research and development expenses incurred under this collaborative arrangement totaled $0.5 million, $0.6 million, and less than $0.1 million for the years ended December 31, 2022, 2021, and 2020, respectively.
3. Recently adopted accounting standards, recently issued accounting pronouncements, and recent law changes
Recently Adopted Accounting Standards
Adoption of Accounting Standards Update (“ASU”) 2021-10—Government Assistance (Topic 832): Disclosures by Business Entities about Government Assistance
In November 2021, the Financial Accounting Standards Board (“FASB”) issued ASU 2021-10, which aims to increase the transparency of government assistance by requiring entities to provide information about the nature of the transaction, terms and conditions associated with the transaction, and financial statement line items affected by the transaction. The Company voluntarily elected to early adopt this standard for the year ended December 31, 2021, on a prospective basis. Adoption of this standard did not have a significant impact to the existing disclosures made in relation to government assistance received by the Company in 2020 as part of the Coronavirus Aid, Relief, and Economic Security Act ("CARES Act").
Adoption of ASU 2019-12, Simplifying the accounting for income taxes
In December 2019, the FASB issued ASU 2019-12, which reduces the complexity of accounting for income taxes by eliminating certain exceptions to the general principles in ASC 740, Income Taxes. Additionally, the ASU simplifies U.S. GAAP by amending the requirements related to the accounting for "hybrid" tax regimes and also adding the requirement to evaluate when a step up in the tax basis of goodwill should be considered part of the business combination and when it should be considered a separate transaction. The Company adopted this ASU effective January 1, 2021, with certain provisions applied retrospectively and other provisions applied prospectively. Adoption of this ASU did not have a material impact to the Company’s condensed consolidated balance sheet, statements of operations, or cash flows.
Adoption of ASU 2016-13, Financial Instruments—Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments and Subsequent Amendments
In June 2016, the FASB issued ASU 2016-13 (which was then further clarified in subsequent ASUs), which required that credit losses for certain types of financial instruments, including accounts receivable, be estimated based on expected credit losses among other changes. The Company adopted this ASU effective as of January 1, 2020, using a modified retrospective approach. See Note 15 for additional discussion of the Company’s adoption of Topic 326 and its resulting accounting policies.
Adoption of ASU 2017-04, Intangibles—Goodwill and Other (Topic 350): Simplifying the Test for Goodwill Impairment
In January 2017, the FASB issued ASU 2017-04, which eliminated Step 2 of the previous goodwill impairment test, which required a hypothetical purchase price allocation to measure goodwill impairment. Under ASU 2017-04, a goodwill impairment loss is now measured as the amount by which a reporting unit’s carrying value exceeds its fair value, not to exceed the recorded amount of goodwill. The Company adopted this ASU effective January 1, 2020, on a prospective basis and followed this guidance to measure the goodwill impairment of $11.8 million recorded in the year ended December 31, 2021.
F-10
Adoption of ASU 2018-13, Fair Value Measurement (Topic 820): Disclosure Framework—Changes to the Disclosure Requirements for Fair Value Measurement
In August 2018, the FASB issued ASU 2018-13, which eliminated certain disclosures, such as the amount and reasons for transfers between Level 1 and Level 2 of the fair value hierarchy, and added new disclosure requirements for Level 3 measurements. The Company adopted this ASU effective January 1, 2020, with certain provisions of the ASU applied retrospectively and other provisions provided prospectively. Adoption of this ASU did not impact the Company’s condensed consolidated balance sheet, statements of operations, or cash flows; however, adoption of the ASU did result in modified disclosures in Note 12.
Adoption of ASU 2018-15, Intangibles—Goodwill and Other—Internal-Use Software (Subtopic 350-40): Customer’s Accounting for Implementation Costs Incurred in a Cloud Computing Arrangement That Is a Service Contract
In August 2018, the FASB issued ASU 2018-15, which aligned the requirements for capitalizing implementation costs incurred in a hosting arrangement that is a service contract with the requirements for capitalizing implementation costs incurred to develop or obtain internal-use software. The accounting for the service element of a hosting arrangement that is a service contract was not affected by the amendments in this update. The Company adopted this ASU effective January 1, 2020, on a prospective basis. Adoption of this ASU did not have a material impact to the Company’s condensed consolidated balance sheet, statements of operations, or cash flows, but is expected to impact future cloud computing arrangements.
Adoption of ASU 2020-04, Reference Rate Reform (Topic 848)
In March 2020, the FASB issued ASU 2020-04, which provided temporary optional guidance to ease the potential financial reporting burden of the expected market transition away from the London Inter-Bank Offered Rate. The new guidance provided optional expedients and exceptions for applying U.S. GAAP to contract modifications, hedge accounting, and other transactions affected by reference rate reform if certain criteria are met through December 31, 2022. The Company adopted this ASU effective March 12, 2020, the effective date of the ASU, on a prospective basis. Adoption of this ASU did not have a material impact to the Company’s condensed consolidated balance sheet, statements of operations, or cash flows.
Recently Issued Accounting Pronouncements
| | | | | | |
Topic | | Description of Guidance | | Effective Date | | Status of Company's Evaluation |
Accounting for Contract Assets and Contract Liabilities from Contracts with Customers (ASU 2021-08) | | Requires that an acquirer recognize and measure contract assets and liabilities acquired in a business combination in accordance with Topic 606, which governs the accounting for revenue contracts with customers. The guidance is to be applied prospectively to acquisitions occurring on or after the effective date, with early adoption permitted. | | January 1, 2023 | | The Company is currently evaluating the impact this ASU may have on its consolidated financial statements. |
Fair Value Measurement of Equity Securities Subject to Contractual Sale Restrictions (ASU 2022-03) | | Clarifies the guidance in Topic 820, Fair Value Measurement, when measuring the fair value of an equity security subject to contractual restrictions that prohibit the sale of an equity security and introduces new disclosure requirements for equity securities subject to contractual sale restrictions. Certain of the provisions are to be applied retrospectively with other provisions applied prospectively. | | January 1, 2024 | | The Company is currently evaluating the impact this ASU may have on its consolidated financial statements. |
Recent Law Changes
COVID-19 and the Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”)
In March 2020, the CARES Act entered into federal law, which was aimed at providing emergency assistance and health care for individuals, families, and businesses affected by the Coronavirus Disease 2019 ("COVID-19") pandemic and to provide general support to the U.S. economy. The CARES Act, among other things, included provisions relating to the deferment of employer side social security payments and technical corrections to tax depreciation methods for qualified improvement property. The CARES Act had no impact to the Company’s income tax expense/benefit reported within the consolidated statements of operations for each of
F-11
the years ended December 31, 2021 and 2020. The CARES Act also provided financial relief to the Company through other various programs, each of which are described in further detail below.
In April 2020, the Company received $13.9 million in funds from the Centers for Medicare & Medicaid Services (“CMS”) Accelerated and Advance Payment Program. For discussion of the Company’s accounting for these funds, see Note 15.
In April 2020, the Company also automatically received $4.7 million in funds from the U.S. Department of Health and Human Services as part of the Provider Relief Fund. The Company recognized this in-substance grant within other income for the year ended December 31, 2020.
In addition, as part of the CARES Act, the Company was permitted to defer all employer social security payroll tax payments for the remainder of the 2020 calendar year subsequent to the CARES Act being signed into federal law, such that 50% of the taxes could be deferred until December 31, 2021, with the remaining 50% deferred until December 31, 2022. As of December 31, 2020, the Company had deferred $0.6 million associated with this program, which was then voluntarily repaid, in full, in 2021.
Consolidated Appropriations Act of 2021 (the “Consolidated Appropriations Act”)
On December 27, 2020, the Consolidated Appropriations Act entered into federal law. The Consolidated Appropriations Act did not have a material impact to the Company’s income tax provision for the year ended December 31, 2021.
American Rescue Plan Act of 2021 (“the American Rescue Plan”)
On March 11, 2021, the American Rescue Plan entered into federal law. The American Rescue Plan, among other things, included provisions related to the deduction of executive compensation beginning in 2027. The American Rescue Plan had no impact to the Company’s condensed consolidated financial statement for the year ended December 31, 2021.
4. Acquisitions
FITBONE Asset Purchase Agreement
In March 2020, the Company completed an Asset Purchase Agreement (the “Purchase Agreement”) with Wittenstein SE (“Wittenstein”), a privately-held German-based company, to acquire assets associated with the FITBONE intramedullary lengthening system for limb lengthening of the femur and tibia bones for $18.0 million in cash consideration. The Company also entered into a Contract Manufacturing and Supply Agreement (“CMSA”) with Wittenstein, which was accounted for as a finance lease.
Distributor Acquisition
In July 2020, the Company acquired certain assets of a medical device distributor for consideration of up to $7.6 million.
Purchase Price Allocations for Completed Acquisitions Discussed Above
| | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | FITBONE | | | Assigned
Useful
Life | | Distributor Acquisition | | | Assigned
Useful
Life |
Assets acquired | | | | | | | | | | |
Inventories | | $ | 528 | | | | | $ | — | | | |
Other long-term assets | | | — | | | | | | — | | | |
Intangible assets | | | | | | | | | | |
Customer relationships | | | 800 | | | 15 years | | | 7,340 | | | 5 years |
Developed technology | | | 4,500 | | | 8 years | | | — | | | N/A |
In-process research and development ("IPR&D") | | | 300 | | | Indefinite | | | — | | | N/A |
Trade name | | | 600 | | | 15 years | | | — | | | N/A |
Assembled workforce | | | — | | | N/A | | | 235 | | | 5 years |
Total identifiable assets acquired | | $ | 6,728 | | | | | $ | 7,575 | | | |
Liabilities assumed | | | — | | | | | | — | | | |
Goodwill | | | 11,272 | | | | | | — | | | |
Total fair value of consideration transferred | | $ | 18,000 | | | | | $ | 7,575 | | | |
F-12
5. Inventories
Inventories are valued at the lower of cost or estimated net realizable value, after provision for excess, obsolete or impaired items, which is reviewed and updated on a periodic basis by management. For inventory procured or produced, whether internally or through contract manufacturing arrangements, at the Company's manufacturing facility in Italy, cost is determined on a weighted-average basis, which approximates the first-in, first-out (“FIFO”) method. For inventory procured or produced, whether internally or through contract manufacturing arrangements, at the Company's manufacturing facilities in Texas and California, standard cost, which approximates actual cost on the FIFO method, is used to value inventory. Standard costs are reviewed by management, at least annually or more often, in the event circumstances indicate a change in cost has occurred.
Work-in-process and finished products include material, labor, and production overhead costs. Field and consignment inventory, which represents immediately saleable finished products inventory that is in the possession of the Company’s independent sales representatives or located at third-party customers, such as distributors and hospitals, is included within finished products. Inventory previously reported as field/consignment inventory has been reclassified to finished products to conform with current period presentation.
| | | | | | | | |
| | December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
Raw materials | | $ | 17,035 | | | $ | 9,589 | |
Work-in-process | | | 19,243 | | | | 15,096 | |
Finished products | | | 63,872 | | | | 58,289 | |
Inventories | | $ | 100,150 | | | $ | 82,974 | |
The Company adjusts the value of its inventory to the extent management determines that the cost cannot be recovered due to obsolescence or other factors. In order to make these determinations, management uses estimates of future demand for each product to determine the appropriate inventory reserves and to make corresponding adjustments to the carrying value of these inventories to reflect the lower of cost or estimated net realizable value.
6. Property, plant, and equipment
Property, plant, and equipment is stated at cost or estimated fair value when acquired as part of a business combination, less accumulated depreciation. Costs include all expenditures necessary to place the asset in service, generally including freight and sales and use taxes. Property, plant, and equipment includes instrumentation held by customers, which is generally used to facilitate the implantation of the Company’s products.
The useful lives of these assets are generally as follows:
| | |
| | Years |
Buildings | | 25 to 33 |
Plant and equipment | | 1 to 10 |
Instrumentation | | 3 to 4 |
Computer software | | 3 to 7 |
Furniture and fixtures | | 4 to 8 |
The Company evaluates the useful lives of these assets on an annual basis. Depreciation is computed on a straight-line basis over the useful lives of the assets. Depreciation of leasehold improvements is computed over the shorter of the lease term or the useful life of the asset. Total depreciation expense was $19.6 million, $20.2 million, and $19.3 million for the years ended December 31, 2022, 2021, and 2020, respectively.
Expenditures for maintenance and repairs and minor renewals and improvements, which do not extend the lives of the respective assets, are expensed as incurred. All other expenditures for renewals and improvements are capitalized. The assets and related accumulated depreciation are adjusted for property retirements and disposals, with the resulting gain or loss included in earnings. Fully depreciated assets remain in the accounts until retired from service.
F-13
| | | | | | | | |
| | December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
Cost | | | | | | |
Buildings | | $ | 3,867 | | | $ | 3,925 | |
Plant and equipment | | | 48,358 | | | | 50,275 | |
Instrumentation | | | 92,607 | | | | 100,515 | |
Computer software | | | 40,685 | | | | 53,200 | |
Furniture and fixtures | | | 7,917 | | | | 8,307 | |
Construction in progress | | | 4,515 | | | | 2,597 | |
Finance lease assets | | | 23,276 | | | | 23,397 | |
Property, plant, and equipment, gross | | | 221,225 | | | | 242,216 | |
Accumulated depreciation | | | (162,996 | ) | | | (182,964 | ) |
Property, plant, and equipment, net | | $ | 58,229 | | | $ | 59,252 | |
The Company capitalizes system development costs related to internal-use software during the application development stage. Costs related to preliminary project activities and post-implementation activities are expensed as incurred. Internal-use software is amortized on a straight-line basis over its estimated useful life, which generally ranges from three to seven years.
Long-lived assets are evaluated for impairment annually or whenever events or changes in circumstances have occurred that would indicate impairment. For purposes of the evaluation, the Company groups its long-lived assets with other assets and liabilities at the lowest level of identifiable cash flows if the asset does not generate cash flows independent of other assets and liabilities. If the carrying value of the asset or asset group exceeds the undiscounted cash flows expected to result from the use and eventual disposition of the asset group, the Company will write the carrying value down to fair value in the period identified.
The Company generally determines fair value of long-lived assets as the present value of estimated future cash flows. In determining the estimated future cash flows associated with the assets, the Company uses estimates and assumptions about future revenue contributions, cost structures, and remaining useful lives of the asset group. The use of alternative assumptions, including estimated cash flows, discount rates, and alternative estimated remaining useful lives could result in different calculations of impairment.
7. Intangible assets
Intangible assets are recorded at cost, or when acquired as a part of a business combination, at estimated fair value, less accumulated amortization. These assets are amortized on a straight-line basis over the useful lives of the assets, which the Company believes is materially consistent with the pattern of economic benefit provided by the assets.
| | | | | | | | | | |
| | | | December 31, | |
(U.S. Dollars, in thousands) | | Weighted Average Amortization Period | | 2022 | | | 2021 | |
Cost | | | | | | | | |
Patents | | 10.0 years | | $ | 40,108 | | | $ | 44,561 | |
Developed technology | | 9.8 years | | | 43,699 | | | | 43,979 | |
IPR&D | | Indefinite | | | 300 | | | | 300 | |
Customer relationships | | 7.8 years | | | 15,572 | | | | 15,621 | |
License and other | | 9.3 years | | | 23,295 | | | | 18,924 | |
Trademarks—finite lived | | 10.0 years | | | 1,875 | | | | 1,839 | |
| | 9.3 years | | | 124,849 | | | | 125,224 | |
Accumulated amortization | | | | | | | | |
Patents | | | | $ | (37,506 | ) | | $ | (41,408 | ) |
Developed technology | | | | | (17,830 | ) | | | (13,409 | ) |
Customer relationships | | | | | (6,938 | ) | | | (4,520 | ) |
License and other | | | | | (14,386 | ) | | | (12,528 | ) |
Trademarks—finite lived | | | | | (801 | ) | | | (693 | ) |
| | | | | (77,461 | ) | | | (72,558 | ) |
Intangible assets, net | | | | $ | 47,388 | | | $ | 52,666 | |
F-14
Acquired IPR&D represents the fair value assigned to acquired research and development assets that have not reached technological feasibility. In a business combination, the fair value assigned to acquired IPR&D is determined by estimating the remaining costs to develop the acquired technology into commercially viable products, estimating the resulting revenues from the projects, and discounting the net cash flows to present value. The revenue and cost projections used to value acquired IPR&D are, as applicable, reduced based on the probability of success of developing the asset. Additionally, estimated revenues consider the relevant market sizes and growth factors, expected trends in technology, and the nature and expected timing of new product introductions by the Company and its competitors. The rates utilized to discount the net cash flows to their present value are commensurate with the stage of development of the project and uncertainties in the economic estimates used in the projections. Any future costs to further develop the IPR&D subsequent to acquisition are recorded to research and development expense as incurred.
IPR&D assets are considered to be indefinite-lived assets until the completion or abandonment of the associated research and development efforts. During the period the assets are considered indefinite-lived, they are not amortized but tested for impairment. Impairment testing is performed at least annually or when a triggering event occurs that could indicate a potential impairment. If and when development is complete, which generally occurs when regulatory approval to market a product is obtained, the associated assets are reclassified to developed technology and are amortized over an assigned useful life that best reflects the economic benefits provided by these assets.
Amortization expense for intangible assets was $9.4 million, $9.4 million, and $11.2 million for the years ended December 31, 2022, December 31, 2021, and 2020, respectively. Future amortization expense for intangible assets is estimated as follows:
| | | | |
(U.S. Dollars, in thousands) | | Amortization | |
2023 | | $ | 9,250 | |
2024 | | | 8,705 | |
2025 | | | 7,692 | |
2026 | | | 6,658 | |
2027 | | | 6,470 | |
Thereafter | | | 8,313 | |
Total finite-lived intangible assets, net | | $ | 47,088 | |
Indefinite-lived intangible assets, net | | | 300 | |
Intangible assets, net | | $ | 47,388 | |
8. Goodwill
The Company tests goodwill at least annually for impairment. The Company tests more frequently if indicators are present or changes in circumstances suggest that impairment may exist. These indicators include, among others, declines in sales, earnings or cash flows, or the development of a material adverse change in the business climate. The Company assesses goodwill for impairment at the reporting unit level, which is defined as an operating segment or one level below an operating segment.
The following table presents the net carrying value of goodwill as of December 31, 2022, and 2021, and a rollforward of such balances from December 31, 2021, by reportable segment:
| | | | | | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | December 31, 2021 | | | Impairment | | | Currency Translation Adjustment | | | December 31, 2022 | |
Global Spine | | $ | 71,317 | | | $ | — | | | $ | — | | | $ | 71,317 | |
Global Orthopedics | | | 11,822 | | | | | | | (692 | ) | | | 11,130 | |
Goodwill, gross | | $ | 83,139 | | | $ | — | | | $ | (692 | ) | | $ | 82,447 | |
Accumulated impairment loss | | | (11,822 | ) | | | — | | | | 692 | | | | (11,130 | ) |
Goodwill, net of accumulated impairment losses | | $ | 71,317 | | | $ | — | | | $ | — | | | $ | 71,317 | |
In the fourth quarter of 2021, the Company performed a quantitative assessment of its goodwill. The Company estimated the fair value of each reporting unit using a weighted average of fair value derived from both an income approach and a market approach (all Level 3 fair value measurements). Upon estimating the fair value of each of its reporting units, the Company determined its Global Orthopedics reporting unit’s fair value was less than its carrying value of net assets. This resulted in recording a full impairment of the Global Orthopedics goodwill of $11.8 million, which was reflected within Acquisition-related amortization and
F-15
remeasurement. This amount also represents the total of the Company’s accumulated goodwill impairment losses as of December 31, 2022, and 2021, respectively. The assessment concluded there were no indicators of impairment for the Global Spine goodwill.
In the fourth quarter of 2022, the Company performed a qualitative assessment for its annual goodwill impairment analysis, which did not result in impairment. This qualitative analysis considered all relevant factors specific to the reporting units, including macroeconomic conditions, industry and market considerations, overall financial performance, and relevant entity-specific events.
9. Leases
The Company determines if a contractual arrangement qualifies as a lease at inception. The Company’s leases primarily relate to facilities, vehicles, equipment, and certain contract manufacturing agreements. Lease assets represent the Company’s right to use an underlying asset for the lease term, while lease liabilities represent the obligation to make lease payments arising from the lease. Lease assets and liabilities are recognized at the commencement date based on the present value of lease payments over the lease term. As the Company’s leases do not provide an implicit rate, the Company’s incremental borrowing rate is used as a discount rate, based on the information available at the commencement date, in determining the present value of lease payments. Lease assets also include the impact of any prepayments made and are reduced by the impact of any lease incentives.
The Company does not recognize lease liabilities or lease assets on the balance sheet for short-term leases (leases with a lease term of twelve months or less as of the commencement date). Rather, any short-term lease payments are recognized as an expense on a straight-line basis over the lease term. The current period short-term lease expense reasonably reflects our short-term lease commitments.
For all classifications of leases, the Company combines lease and non-lease components to account for them as a single lease component. Variable lease payments are excluded from the lease liability and recognized in the period in which the obligation is incurred. Additionally, lease terms may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise the option.
A summary of the Company’s lease portfolio as of December 31, 2022, and 2021, is presented in the table below:
| | | | | | | | | | |
(U.S. Dollars, in thousands, except lease term and discount rate) | | Classification | | December 31, 2022 | | | December 31, 2021 | |
Assets | | | | | | | | |
Operating leases | | Other long-term assets | | $ | 6,788 | | | $ | 3,155 | |
Finance leases | | Property, plant and equipment, net | | | 17,360 | | | | 18,600 | |
Total lease assets | | | | $ | 24,148 | | | $ | 21,755 | |
Liabilities | | | | | | | | |
Current | | | | | | | | |
Operating leases | | Other current liabilities | | $ | 1,638 | | | $ | 1,834 | |
Finance leases | | Current portion of finance lease liability | | | 652 | | | | 2,590 | |
Long-term | | | | | | | | |
Operating leases | | Other long-term liabilities | | | 5,376 | | | | 1,443 | |
Finance leases | | Long-term portion of finance lease liability | | | 19,239 | | | | 19,890 | |
Total lease liabilities | | | | $ | 26,905 | | | $ | 25,757 | |
| | | | | | | | |
Weighted Average Remaining Lease Term | | | | | | | | |
Operating leases | | | | 4.5 years | | | 3.3 years | |
Finance leases | | | | 17.6 years | | | 17.0 years | |
| | | | | | | | |
Weighted Average Discount Rate | | | | | | | | |
Operating leases | | | | | 4.0 | % | | | 2.6 | % |
Finance leases | | | | | 4.4 | % | | | 4.2 | % |
F-16
The components of lease costs were as follows:
| | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | For the Year Ended December 31, 2022 | | | For the Year Ended December 31, 2021 | | | For the Year Ended December 31, 2020 | |
Finance lease costs: | | | | | | | | | |
Amortization of right-of-use assets | | $ | 1,238 | | | $ | 2,049 | | | $ | 1,766 | |
Interest on finance lease liabilities | | | 890 | | | | 933 | | | | 940 | |
Operating lease costs | | | 2,126 | | | | 2,234 | | | | 2,235 | |
Short-term lease costs | | | 152 | | | | 213 | | | | 230 | |
Variable lease costs | | | 932 | | | | 815 | | | | 673 | |
Total lease costs | | $ | 5,338 | | | $ | 6,244 | | | $ | 5,844 | |
Supplemental cash flow information related to leases was as follows:
| | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | For the Year Ended December 31, 2022 | | | For the Year Ended December 31, 2021 | | | For the Year Ended December 31, 2020 | |
Cash paid for amounts included in the measurement of lease liabilities | | | | | | | | | |
Operating cash flows from operating leases | | $ | 3,805 | | | $ | 4,627 | | | $ | 4,299 | |
Operating cash flows from finance leases | | | 885 | | | | 907 | | | | 689 | |
Financing cash flows from finance leases | | | 2,594 | | | | 537 | | | | 323 | |
Right-of-use assets obtained in exchange for lease obligations | | | | | | | | | |
Operating leases | | | 5,603 | | | | 589 | | | | 959 | |
Finance leases | | | — | | | | 149 | | | | 1,949 | |
A summary of the Company’s remaining lease liabilities as of December 31, 2022, is included below:
| | | | | | | | |
(U.S. Dollars, in thousands) | | Operating
Leases | | | Finance
Leases | |
2023 | | $ | 1,821 | | | $ | 1,508 | |
2024 | | | 1,587 | | | | 1,538 | |
2025 | | | 1,497 | | | | 1,543 | |
2026 | | | 1,411 | | | | 1,562 | |
2027 | | | 1,211 | | | | 1,593 | |
Thereafter | | | 129 | | | | 21,021 | |
Total undiscounted value of lease liabilities | | | 7,656 | | | | 28,765 | |
Less: Interest | | | (642 | ) | | | (8,874 | ) |
Present value of lease liabilities | | $ | 7,014 | | | $ | 19,891 | |
| | | | | | |
Current portion of lease liabilities | | $ | 1,638 | | | $ | 652 | |
Long-term portion of lease liabilities | | | 5,376 | | | | 19,239 | |
Total lease liabilities | | $ | 7,014 | | | $ | 19,891 | |
F-17
10. Other current liabilities
| | | | | | | | |
| | December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
Accrued expenses | | $ | 9,611 | | | $ | 7,151 | |
Salaries, bonuses, commissions, and related taxes payable | | | 18,531 | | | | 23,552 | |
Accrued distributor commissions | | | 10,483 | | | | 10,787 | |
Accrued legal and settlement expenses | | | 3,891 | | | | 3,794 | |
Contingent consideration liability | | | 1,000 | | | | 17,200 | |
Short-term operating lease liability | | | 1,638 | | | | 1,834 | |
Non-income taxes payable | | | 6,586 | | | | 4,655 | |
Accelerated and advance payment program | | | — | | | | 4,791 | |
Other payables | | | 3,634 | | | | 3,017 | |
Other current liabilities | | $ | 55,374 | | | $ | 76,781 | |
11. Long-term debt
On October 25, 2019, the Company, and certain of its wholly-owned subsidiaries (collectively with the Company, the “Borrowers”), as borrowers, and certain material subsidiaries of the Company as guarantors, entered into a Second Amended and Restated Credit Agreement (the “Amended Credit Agreement”) with JPMorgan Chase Bank, N.A. (“JPMorgan”), as Administrative Agent, and certain lender parties thereto. The Amended Credit Agreement provides for a $300.0 million secured revolving credit facility (the “Facility”) amending and restating the $125.0 million secured revolving credit facility that previously existed with such lenders. The Credit Agreement has a maturity date of October 25, 2024. On March 1, 2023, the Amended Credit Agreement and the Facility were amended to replace London Inter-Bank Offered Rate ("LIBOR")-based pricing with Secured Overnight Financing Rate ("SOFR")-based pricing.
In April 2020, as a precautionary measure to increase the Company’s cash position and to preserve financial flexibility during the initial uncertainty resulting from the COVID-19 pandemic, the Company completed a borrowing of $100.0 million under the Facility, which the Company then paid back in full later that year. The Company had no borrowings outstanding under the Facility at December 31, 2022, and 2021, respectively. However, on January 3, 2023, the Company borrowed $30.0 million under the Facility for working capital purposes, including to fund certain merger-related expenses. Further, an additional $15.0 million was borrowed on March 3, 2023.
Borrowings under the Amended Credit Agreement may be used for, among other things, working capital and other general corporate purposes of the Company and its subsidiaries (including permitted acquisitions and permitted payments of dividends and other distributions). The Facility is available in U.S. Dollars with up to $150.0 million of the Facility available to be borrowed in Euros or Pound Sterling (the “Agreed Currencies”). The Facility further permits up to $50.0 million of the Facility to be utilized for the issuance of letters of credit in the Agreed Currencies. The Borrowers have the ability to increase the amount of the Facility, which increases may take the form of increases to the revolving credit commitments or the issuance of new term A loans, by an aggregate amount of up to the greater of $150.0 million or an incremental amount such that the total amount of the Facility does not exceed 350% of consolidated EBITDA of the Company (as determined for the four fiscal quarter period most recently ended for which financial statements are available), upon satisfaction of customary conditions precedent for such increases or incremental loans and receipt of additional commitments by one or more existing or new lenders.
Borrowings under the Facility bear interest at a floating rate, which is, at the Borrowers’ option, either SOFR, plus an applicable margin ranging from 1.25% to 2.25% or a base rate plus an applicable margin ranging from 0.25% to 1.25% (in each case subject to adjustment based on the Company’s total leverage ratio). An unused fee ranging from 0.15% to 0.25% (subject to adjustment based on the Company’s total leverage ratio) is payable quarterly in arrears based on the daily amount of the undrawn portion of each lender’s revolving credit commitment under the Facility. Fees are payable on outstanding letters of credit at a rate equal to the applicable margin for SOFR loans, plus certain customary fees payable solely to the issuer of the letter of credit.
Certain of the Company’s existing and future material subsidiaries (collectively, the “Guarantors”) are required to guarantee the repayment of the Borrowers’ obligations under the Amended Credit Agreement. The obligations of the Borrowers and each of the Guarantors with respect to the Amended Credit Agreement are secured by a pledge of substantially all of the personal property
F-18
assets of the Borrowers and each of the Guarantors, including accounts receivables, deposit accounts, intellectual property, investment property, inventory, equipment, and equity interests in their respective subsidiaries.
The Amended Credit Agreement contains customary affirmative and negative covenants, including limitations on the Company’s ability to incur additional debt, grant or permit additional liens, make investments and acquisitions, merge or consolidate with others, dispose of assets, pay dividends and distributions, pay subordinated indebtedness, and enter into affiliate transactions. In addition, the Amended Credit Agreement contains financial covenants requiring the Company on a consolidated basis to maintain, as of the last day of any fiscal quarter, a total net leverage ratio of not more than 3.5 to 1.0 (which ratio can be permitted to increase to 4.0 to 1.0 for no more than 4 fiscal quarters following a material acquisition) and an interest coverage ratio of at least 3.0 to 1.0. The Amended Credit Agreement also includes events of default customary for facilities of this type and upon the occurrence of such events of default, subject to customary cure rights, all outstanding loans under the Facility may be accelerated and/or the lenders’ commitments terminated. The Company is in compliance with all required financial covenants as of December 31, 2022.
In conjunction with obtaining the Facility, the Company paid $1.5 million in debt issuance costs and capitalized a total of $1.8 million associated with the Facility (inclusive of certain capitalized costs prior to the most recent amendment). These costs are being amortized over the life of the Facility. Capitalized debt issuance costs are included in other long-term assets, net of accumulated amortization. As of December 31, 2022, and December 31, 2021, debt issuance costs, net of accumulated amortization, were $0.7 million and $1.0 million, respectively. Debt issuance costs amortized or expensed totaled $0.4 million for each of the years ended December 31, 2022, 2021, and 2020, respectively.
The Company has an unused available Italian line of credit of €5.5 million ($5.9 million and $6.3 million) at December 31, 2022, and 2021, respectively. This unsecured line of credit provides the Company the option to borrow amounts in Italy at interest rates determined at the time of borrowing.
The Company paid cash related to interest of $1.4 million, $1.5 million, and $1.9 million for the years ended December 31, 2022, 2021, and 2020, respectively.
12. Fair value measurements and investments
Fair value is defined as the price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Non-financial assets and liabilities of the Company measured at fair value include any long-lived assets that are impaired in a currently reported period or equity securities measured at observable prices in orderly transactions. The authoritative guidance also describes three levels of inputs that may be used to measure fair value:
| |
Level 1: | quoted prices in active markets for identical assets and liabilities |
| |
Level 2: | observable inputs other than quoted prices in active markets for identical assets and liabilities |
| |
Level 3: | unobservable inputs in which there is little or no market data available, which require the reporting entity to develop its own assumptions |
The Company’s financial instruments include cash equivalents, accounts receivable, accounts payable, long-term secured debt, available for sale debt securities, equity securities, contingent consideration, and deferred compensation plan liabilities. The carrying value of cash equivalents, accounts receivable, and accounts payable approximate fair value due to the short-term maturities of these instruments. The Company’s secured revolving credit facility carries a floating rate of interest; therefore, the carrying value of long-term debt is considered to approximate the fair value.
F-19
The Company’s available for sale debt securities, equity securities, contingent consideration, and deferred compensation plan liabilities are the only financial instruments recorded at fair value on a recurring basis as follows:
| | | | | | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | Balance
December 31,
2022 | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets | | | | | | | | | | | | |
Neo Medical convertible loan agreement | | $ | 7,140 | | | $ | — | | | $ | — | | | $ | 7,140 | |
Neo Medical preferred equity securities | | | 6,084 | | | | — | | | | 6,084 | | | | — | |
Bone Biologics equity securities | | | — | | | | — | | | | — | | | | — | |
Other investments | | | 1,726 | | | | — | | | | — | | | | 1,726 | |
Total | | $ | 14,950 | | | $ | — | | | $ | 6,084 | | | $ | 8,866 | |
Liabilities | | | | | | | | | | | | |
Spinal Kinetics contingent consideration | | | — | | | $ | — | | | $ | — | | | $ | — | |
Deferred compensation plan | | | (1,515 | ) | | | — | | | | (1,515 | ) | | | — | |
Total | | $ | (1,515 | ) | | $ | — | | | $ | (1,515 | ) | | $ | — | |
| | | | | | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | Balance
December 31,
2021 | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets | | | | | | | | | | | | |
Neo Medical convertible loan agreements | | $ | 7,148 | | | $ | — | | | $ | — | | | $ | 7,148 | |
Neo Medical preferred equity securities | | | 5,413 | | | | — | | | | 5,413 | | | | — | |
Bone Biologics equity securities | | | 309 | | | | 309 | | | | — | | | | — | |
Other Investments | | | 1,505 | | | | — | | | | — | | | | 1,505 | |
Total | | $ | 14,375 | | | $ | 309 | | | $ | 5,413 | | | $ | 7,148 | |
Liabilities | | | | | | | | | | | | |
Spinal Kinetics contingent consideration | | $ | (17,200 | ) | | $ | — | | | $ | — | | | $ | (17,200 | ) |
Deferred compensation plan | | | (1,314 | ) | | | — | | | | (1,314 | ) | | | — | |
Total | | $ | (18,514 | ) | | $ | — | | | $ | (1,314 | ) | | $ | (17,200 | ) |
The fair value of the Company’s deferred compensation plan liabilities are determined based on inputs that are readily available in public markets or that can be derived from information available in publicly quoted markets; therefore, the Company has categorized this liability as a Level 2 financial instrument.
Neo Medical Convertible Loan Agreements and Equity Investment
On October 1, 2020, the Company purchased shares of Neo Medical’s preferred stock for consideration of $5.0 million and entered into a Convertible Loan Agreement pursuant to which Orthofix loaned Neo Medical CHF 4.6 million, or $5.0 million at the date of issuance (the “Convertible Loan”). The loan bears interest at 8.0%, with interest due semi-annually. At each interest payment date, the borrower may elect to capitalize any interest due to the then outstanding principal balance of the loan. The Convertible Loan matures on October 1, 2024. If a change in control of Neo Medical occurs prior to the maturity date, the Convertible Loan shall become immediately due upon such event. The Convertible Loan may be convertible by either party into shares of Neo Medical’s preferred stock. The Company may convert the loan at its own election at any time prior to the full repayment or settlement of the Convertible Loan. Neo Medical may elect to convert the loan only in the event of a qualified financing event, as defined within the agreement. The price per share at which the loan converts is dependent upon i) the party electing conversion and ii) Neo Medical’s price per share in its most recent fundraising activities at the time of conversion, as specified within the agreement.
In October 2021, the Company entered into an additional Convertible Loan Agreement (the “Additional Convertible Loan”), pursuant to which the Company loaned Neo Medical an additional CHF 0.6 million ($0.7 million as of the issuance date). In January 2022, the Company elected to convert the Additional Convertible Loan into shares of Neo Medical’s preferred stock.
The equity securities are recorded in other long-term assets and are considered an investment that does not have a readily determinable fair value. As such, the Company measures this investment at cost, less any impairment, plus or minus changes resulting from observable price changes in orderly transactions for identical or similar investments of the same issuer.
The table below presents a reconciliation of the carrying value of the Company’s investment in Neo Medical preferred equity securities for the years ended December 31, 2022, and 2021:
F-20
| | | | | | | | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
Fair value of Neo Medical preferred equity securities at January 1 | | $ | 5,413 | | | $ | 5,000 | |
Conversion of loan into preferred equity securities | | | 671 | | | | — | |
Foreign currency remeasurement recognized in other income, net | | | — | | | | 77 | |
Unrealized gain recognized in other income (expense), net | | | — | | | | 336 | |
Fair value of Neo Medical preferred equity securities at December 31 | | | 6,084 | | | | 5,413 | |
Cumulative unrealized gain on Neo Medical preferred equity securities | | | 413 | | | | 413 | |
The remaining Convertible Loan is recorded in other long-term assets as an available for sale debt security as of December 31, 2022. The Convertible Loan is recorded at fair value, with applicable interest recorded in interest income. The fair value of the Convertible Loan is based upon significant unobservable inputs, including the use of option-pricing models, Monte Carlo simulations for certain periods, and a probability-weighted discounted cash flows model, requiring the Company to develop its own assumptions. Therefore, the Company has categorized this asset as a Level 3 financial asset.
Some of the more significant unobservable inputs used in the fair value measurement of the Convertible Loan include applicable discount rates, implied volatility, the likelihood and projected timing of repayment or conversion, and projected cash flows in support of the estimated enterprise value of Neo Medical. Holding other inputs constant, changes in these assumptions could result in a significant change in the fair value of the Convertible Loan. If the amortized cost of the Convertible Loan exceeds its estimated fair value, the security is deemed to be impaired, and must be evaluated for the recognition of credit losses. Impairment resulting from credit losses is recognized within the statement of income, while impairment resulting from other factors is recognized within other comprehensive income (loss). As of December 31, 2022, the Company has not recognized any credit losses related to the Convertible Loan.
The following table provides a reconciliation of the beginning and ending balances of the Convertible Loan(s), measured at fair value using significant unobservable inputs (Level 3):
| | | | | | | | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
Fair value of Neo Medical Convertible Loans at January 1 | | $ | 7,148 | | | $ | 7,160 | |
Additions | | | — | | | | 671 | |
Interest recognized in interest income, net | | | 436 | | | | 421 | |
Foreign currency remeasurement recognized in other income (expense), net | | | (67 | ) | | | (162 | ) |
Unrealized gain (loss) recognized in other comprehensive income (loss) | | | 294 | | | | (942 | ) |
Conversion of Additional Convertible Loan into preferred equity securities | | | (671 | ) | | | — | |
Fair value of Neo Medical Convertible Loan(s) at December 31 | | | 7,140 | | | | 7,148 | |
Amortized cost basis of Neo Medical Convertible Loan(s) at December 31 | | | 5,907 | | | | 6,209 | |
The following table provides quantitative information related to certain key assumptions utilized within the valuation of the Convertible Loan as of December 31, 2022:
| | | | | | | | | | |
(U.S. Dollars, in thousands) | | Fair Value as of December 31, 2022 | | | Unobservable inputs | | Estimate | |
Neo Medical Convertible Loan | | $ | 7,140 | | | Cost of equity discount rate | | | 18.0 | % |
| | | | | Implied volatility | | | 73.9 | % |
F-21
Bone Biologics Equity Securities
Until August of 2022, the Company held an investment in common stock of Bone Biologics Inc. (“Bone Biologics”), a developer of orthobiologic products. Prior to 2021, the equity securities were considered an investment that did not have a readily determinable fair value as Bone Biologics had very limited trading volumes. As such, the Company measured the investments at cost, less any impairments, plus or minus changes resulting from observable price changes in orderly transactions for an identical or similar investment of the same issuer.
In 2021, Bone Biologics completed a public offering of units, with each unit consisting of one share of common stock and one warrant to purchase common shares. As a result, Bone Biologics’ common stock became actively traded on the NASDAQ (ticker BBLG). The Company concluded the investment represented a Level 1 fair value measurement subsequent to the public offering as the common shares subsequently had quoted prices in active markets for identical assets. As such, the Company recorded the investment at fair value, with changes in fair value recorded within other income (expense), net, subsequent to the public offering.
The following table presents the changes in fair value recognized for each of the years ended December 31, 2022, 2021, and 2020:
| | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Bone Biologics equity securities at January 1 | | $ | 309 | | | $ | — | | | $ | 219 | |
Fair value adjustments and impairments recognized in other income (expense), net | | | (183 | ) | | | 309 | | | | (219 | ) |
Proceeds from the disposition of equity securities | | | (126 | ) | | | — | | | | — | |
Bone Biologics equity securities at December 31 | | $ | — | | | $ | 309 | | | | — | |
Other investments
Other investments represent other assets and investments recorded at fair value that are not deemed to be material for disclosure on an individual basis. The fair value of these assets is based upon significant unobservable inputs, such as probability-weighted discounted cash flows models, requiring the Company to develop its own assumptions. Therefore, the Company has categorized these assets as Level 3 financial assets. As of December 31, 2022, this balance was classified within other current assets, while as of December 31, 2021, this balance was classified within other long-term assets.
Contingent Consideration
The Company recognized a contingent consideration obligation in connection with the acquisition of Spinal Kinetics in 2018. The Spinal Kinetics contingent consideration consists of potential future milestone payments of up to $60.0 million in cash. The milestone payments included (i) $15.0 million upon U.S. Food and Drug Administration (“FDA”) approval of the M6-C artificial cervical disc (the “FDA Milestone”) and (ii) revenue-based milestone payments of up to $45.0 million in connection with future sales of the acquired artificial discs. To trigger applicable payments, milestones must be achieved by April 30, 2023. The FDA Milestone was achieved and paid in 2019. A second milestone payment, totaling $15.0 million, was achieved and paid in 2021 upon meeting certain net sales targets.
The estimated fair value of the remaining Spinal Kinetics contingent consideration, attributable to a revenue-based milestone, was concluded to be zero as of December 31, 2022, as the Company does not expect to achieve the milestone by April 30, 2023. The estimated fair value reflects assumptions made by management as of December 31, 2022, such as the expected timing and volume of elective procedures and the impact of these procedures on future revenues. Any changes in fair value are recorded as an operating expense within acquisition-related amortization and remeasurement.
The following table provides a reconciliation of the beginning and ending balances for the contingent consideration measured at fair value using significant unobservable inputs (Level 3):
| | | | | | | | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
Spinal Kinetics contingent consideration at January 1 | | $ | 17,200 | | | $ | 35,400 | |
Decrease in fair value recognized in acquisition-related amortization and remeasurement | | | (17,200 | ) | | | (3,200 | ) |
Payment made | | | — | | | | (15,000 | ) |
Spinal Kinetics contingent consideration at December 31 | | | — | | | $ | 17,200 | |
F-22
13. Commitments and Contingencies
Contingencies policy
The Company records accruals for certain outstanding legal proceedings, investigations, or claims when it is probable that a liability has been incurred and the amount of the loss can be reasonably estimated. The Company evaluates developments in legal proceedings, investigations, and claims that could affect the amount of any accrual, as well as any developments that would make a loss contingency both probable and reasonably estimable on a quarterly basis. When a loss contingency is not both probable and reasonably estimable, the Company does not accrue the loss. However, if the loss (or an additional loss in excess of the accrual) is at least a reasonable possibility and material, then the Company discloses a reasonable estimate of the possible loss or range of loss, if such reasonable estimate can be made. If the Company cannot make a reasonable estimate of the possible loss, or range of loss, then that is disclosed. In addition, legal fees and other directly related costs are expensed as incurred.
In addition to the matters described in the paragraphs below, in the normal course of its business, the Company is involved in various lawsuits from time to time and may be subject to certain other contingencies. The Company believes any losses related to these matters are individually and collectively immaterial as to a possible loss and range of loss.
Italian Medical Device Payback (“IMDP”)
In 2015, the Italian Parliament introduced rules for entities that supply goods and services to the Italian National Healthcare System. A key provision of the law is a ‘payback’ measure, requiring medical device companies in Italy to make payments to the Italian government if medical device expenditures exceed regional maximum ceilings. Companies are required to make payments equal to a percentage of expenditures exceeding maximum regional caps.
In the third quarter of 2022, the Italian Ministry of Health provided guidelines to the Italian regions and provinces on seeking payback of expenditure overruns relating to the years ended December 31, 2015, through December 31, 2018. Since receiving the guidelines, several regions and provinces have requested payment from affected medical device companies, including the Company. The Company has taken legal action to dispute the legality of such measures.
The Company accounts for the estimated cost of the IMDP as sales and marketing expense and periodically reassesses the liability based upon current facts and circumstances. As a result, the Company recorded expense of $1.2 million for the year ended December 31, 2022, a benefit of $1.2 million for the year ended December 31, 2021, as a result of certain temporary relief provided by the Italian National Healthcare System in response to the COVID-19 pandemic, and expense of $1.5 million for the year ended December 31, 2020. As of December 31, 2022, the Company has accrued $5.9 million related to the IMDP, which it has classified within other long-term liabilities; however, the actual liability could be higher or lower than the amount accrued once all legal proceedings are resolved and upon further clarification of the IMDP by the Italian authorities for more recent fiscal years.
14. Shareholders’ equity
Dividends
The Company has not historically paid dividends to holders of its common stock. Certain subsidiaries of the Company have restrictions on their ability to pay dividends in certain circumstances pursuant to the Amended Credit Agreement. In the event that the Company decides to pay a dividend to holders of its common stock in the future with dividends received from its subsidiaries, the Company may, based on prevailing rates of taxation, be required to pay additional withholding and income tax on such amounts received from its subsidiaries.
F-23
Accumulated Other Comprehensive Income (Loss)
Accumulated other comprehensive income (loss) is comprised of foreign currency translation adjustments and unrealized gains (losses) on available for sale debt securities. The Company’s policy is to release income tax effects related to items recognized within accumulated other comprehensive income (loss) using a portfolio approach. The components of and changes in accumulated other comprehensive income (loss) are as follows:
| | | | | | | | | | | | | | | | |
(U.S. Dollars, in thousands) | | Currency
Translation
Adjustments | | | Neo Medical Convertible Loans | | | Other Investments | | | Accumulated Other
Comprehensive
Income (Loss) | |
Balance at December 31, 2019 | | $ | (3,039 | ) | | $ | — | | | $ | — | | | $ | (3,039 | ) |
Other comprehensive income | | | 4,872 | | | | 1,881 | | | | — | | | | 6,753 | |
Income taxes | | | — | | | | (462 | ) | | | — | | | | (462 | ) |
Balance at December 31, 2020 | | $ | 1,833 | | | $ | 1,419 | | | $ | — | | | $ | 3,252 | |
Other comprehensive loss | | | (2,544 | ) | | | (942 | ) | | | — | | | | (3,486 | ) |
Income taxes | | | — | | | | 234 | | | | — | | | | 234 | |
Balance at December 31, 2021 | | $ | (711 | ) | | $ | 711 | | | $ | — | | | $ | — | |
Other comprehensive income (loss) | | | (1,771 | ) | | | 294 | | | | 101 | | | | (1,376 | ) |
Income taxes | | | — | | | | — | | | | — | | | | — | |
Balance at December 31, 2022 | | $ | (2,482 | ) | | $ | 1,005 | | | $ | 101 | | | $ | (1,376 | ) |
15. Revenue recognition and accounts receivable
Revenue Recognition
The Company accounts for a contract when there is (i) approval and commitment from both parties, (ii) the rights of the parties are identified, (iii) payment terms are identified, (iv) the contract has commercial substance, (v) and collectability of consideration is probable. The Company’s contracts may contain one or more performance obligations. If a contract contains more than one performance obligation, the Company allocates the total transaction price to each of the performance obligations based upon the observable standalone selling price of the promised goods or services underlying each performance obligation. The Company recognizes revenue when control of the promised goods or services is transferred to the customer, which typically occurs at a point in time upon shipment, delivery, or utilization, in an amount that reflects the consideration which the Company expects to be entitled to in exchange for the promised goods or services. The consideration for goods or services reflects any fixed amount stated per the contract and estimates for any variable consideration, such as discounts, to the extent that is it probable that a significant reversal of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is resolved.
The following sections discuss the Company’s revenue recognition policies by significant product category:
Bone Growth Therapies
Bone Growth Therapies revenue is largely attributable to the U.S. and is comprised of third-party payor transactions and wholesale revenue.
The largest portion of Bone Growth Therapies revenue is derived from third-party payors. This includes commercial insurance carriers, health maintenance organizations, preferred provider organizations, and governmental payors, such as Medicare. Revenue is recognized when the product is fitted to and accepted by the patient and all applicable documents required by the third-party payor have been obtained. Amounts paid by third-party payors are generally based on fixed or allowable reimbursement rates. These revenues are recorded at the expected or preauthorized reimbursement rates, net of any contractual allowances or adjustments. Certain billings are subject to review by the third-party payors and may be subject to adjustment.
Wholesale revenue is related to the sale of the Company’s bone growth stimulators directly to durable medical equipment suppliers. Wholesale revenues are typically recognized upon shipment and receipt of a confirming purchase order, which is when the customer obtains control of the promised goods.
F-24
Biologics
Biologics revenue is largely attributable to the U.S. and is primarily related to a collaborative arrangement with MTF, which extends through December 31, 2032. Under this arrangement, the Company markets tissue for bone repair and reconstruction under the brand names Trinity Evolution and Trinity ELITE. Per the terms of the agreement, MTF sources the tissue, processes it to create the allografts, packages, and delivers the tissue to the customer. The Company has exclusive global marketing rights for the Virtuos Lyograft and Trinity ELITE tissue forms, exclusive rights to market FiberFuse Advanced, FiberFuse Strip, and certain other tissues in the U.S., non-exclusive marketing rights for certain other products, and receives marketing fees from MTF based on total sales. MTF is considered the primary obligor in these arrangements; therefore, the Company recognizes marketing service fees on a net basis within net sales upon shipment of the product to the customer and receipt of a confirming purchase order.
Spinal Implants and Global Orthopedics
Spinal Implants and Global Orthopedics products are distributed world-wide, with U.S. sales largely comprised of commercial sales and international sales derived from both commercial sales and stocking distributor arrangements.
Commercial revenue is largely related to the sale of the Company’s Spinal Implants and Global Orthopedics products to hospital customers. The customer obtains control and revenues are recognized when these products have been utilized and a confirming purchase order has been received from the hospital.
Other revenues within the Spinal Implants and Global Orthopedics product categories are derived from stocking distributors, who purchase the Company’s products and then re-sell them directly to customers, such as hospitals. For stocking distributor arrangements, it is the Company’s policy to recognize revenue upon shipment and receipt of a confirming purchase order, which is when the distributor obtains control of the promised goods. The transaction price for revenue recognition is estimated based upon the Company’s historical collection experience with the stocking distributor.
Product Sales and Marketing Service Fees
The table below presents net sales, which includes product sales and marketing service fees, for each of the years ended December 31, 2022, 2021, and 2020.
| | | | | | | | | | | | |
| | For the year ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Product sales | | $ | 405,437 | | | $ | 409,554 | | | $ | 353,087 | |
Marketing service fees | | | 55,276 | | | | 54,925 | | | | 53,475 | |
Net sales | | $ | 460,713 | | | $ | 464,479 | | | $ | 406,562 | |
Product sales primarily consists of the sale of Bone Growth Therapies, Spinal Implants, and Global Orthopedics products. Marketing service fees are received from MTF based on total sales of biologics tissues and relates solely to the Biologics product category within the Global Spine reporting segment. Marketing service fees received from MTF were $55.3 million, or approximately 98% of total Biologics revenues, for the year ended December 31, 2022. As MTF is the single supplier for certain allografts in the Company’s Biologics portfolio, derived from deceased donors for their bone grafts and living donors for their amnion grafts, any event or circumstance that would impact MTF’s continued access to donors or the Company’s ability to market these tissues may adversely impact the Company’s financial results.
Revenues exclude any value added or other local taxes, intercompany sales, and trade discounts. Shipping and handling costs for products shipped to customers are included in cost of sales, and were $4.2 million, $3.5 million, and $2.4 million for the years ended December 31, 2022, 2021, and 2020, respectively.
Accounts receivable and related allowances
Payment terms vary by the type and location of the Company’s customers and the products or services offered. The term between invoicing and when payment is due is not significant.
The Company’s allowance for expected credit losses represents the portion of the receivable’s amortized cost basis that an entity does not expect to collect over the receivable’s contractual life, considering past events, current conditions, and reasonable and supportable forecasts of future economic conditions.
F-25
The process for estimating the ultimate collection of accounts receivable involves certain assumptions and judgments. The determination of the contractual life of accounts receivable, the aging of outstanding receivables, as well as the historical collections, write-offs, and payor reimbursement experience over the estimated contractual lives of such receivables, are integral parts of the estimation process related to reserves for expected credit losses and the establishment of contractual allowances. Accounts receivable are analyzed on a quarterly basis to assess the adequacy of both reserves for expected credit losses and contractual allowances. Revisions in allowances for expected credit loss estimates are recorded as an adjustment to bad debt expense within sales and marketing expenses. Revisions to contractual allowances are recorded as an adjustment to net sales. These estimates are periodically tested against actual collection experience. In addition, the Company analyzes its receivables by geography and by customer type, where appropriate, in developing estimates for expected credit losses.
The following table provides a detail of changes in the Company’s allowance for expected credit losses for the years ended December 31, 2022, and 2021:
| | | | | | | | |
| | For the year ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
Allowance for expected credit losses beginning balance | | $ | 4,944 | | | $ | 4,848 | |
Current period provision for expected credit losses | | | 2,095 | | | | 444 | |
Write-offs charged against the allowance and other | | | (450 | ) | | | (126 | ) |
Effect of changes in foreign exchange rates | | | (170 | ) | | | (222 | ) |
Allowance for expected credit losses ending balance | | $ | 6,419 | | | $ | 4,944 | |
The Company will generally sell receivables from certain Italian public hospitals each year to accelerate cash collections. During 2022, 2021, and 2020, the Company sold €9.2 million, €8.4 million, and €8.3 million ($9.6 million, $9.9 million, and $9.6 million) of receivables, respectively. The related fees for 2022, 2021, and 2020, were $0.3 million, $0.2 million, and $0.3 million, respectively, which were recorded as interest expense. Accounts receivables sold without recourse are removed from the balance sheet at the time of sale.
Contract Liabilities
The Company’s contract liabilities largely relate to a prepayment of $13.9 million received in 2020 from the CMS as part of the Accelerated and Advance Payment Program of the CARES Act.
On October 1, 2020, the President of the United States signed the “Continuing Appropriations Act, 2021 and Other Extensions Act,” which relaxed a number of the Medicare Accelerated and Advance Payment Program’s recoupment terms for providers and suppliers that received funds from the program. In April 2021, Medicare began to recoup 25% of Medicare payments otherwise owed to the provider or supplier for submitted claims. Recoupment then increased to 50% of Medicare payments in March 2022. Thus, during these time periods, rather than receiving the full amount of payment for newly submitted claims, the Company’s outstanding balance under the Accelerated and Advance Payment Program was reduced by the recoupment amount until the full balance had been repaid.
The following table provides a detail of changes in the Company’s contract liability associated with the Accelerated and Advanced Payment Program for the years ended December 31, 2022, and 2021:
| | | | | | | | |
| | For the Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
Contract liability beginning balance | | $ | 4,791 | | | $ | 13,851 | |
Recoupment recognized in net sales | | | (4,791 | ) | | | (9,060 | ) |
Contract liability ending balance | | $ | — | | | $ | 4,791 | |
Other Contract Assets
The Company’s contract assets, excluding accounts receivable (“Other Contract Assets”), largely consist of payments made to certain distributors to obtain contracts, gain access to customers in certain territories, and to provide the benefit of the exclusive distribution of the Company’s products. Other Contract Assets are included in other long-term assets and totaled $1.1 million and $1.4 million as of December 31, 2022, and 2021, respectively.
F-26
Other Contract Assets are amortized on a straight-line basis over the term of the related contract. No impairments were incurred for other contract assets in 2022 or 2021. Further, the Company applies the practical expedient to expense sales commissions when incurred, as the applicable amortization period would be for one year or less.
16. Business segment information
As of December 31, 2022, the Company's operations were managed through two reporting segments: Global Spine and Global Orthopedics. These reporting segments represent the operating segments for which the Chief Executive Officer, who is also Chief Operating Decision Maker (the “CODM”), reviews financial information and makes resource allocation decisions among businesses. As of December 31, 2022, the primary metric used by the CODM in managing the Company is earnings before interest, tax, depreciation, and amortization (“EBITDA”). The Company neither discretely allocates assets, other than goodwill, to its operating segments nor evaluates the operating segments using discrete asset information.
Following the merger with SeaSpine, which was completed on January 5, 2023, the Company expects to reassess its reporting segments in the first quarter of 2023 based on how the operations of the newly combined company will be managed The Company will also reassess its identified segment profitability metric at that time. Accordingly, the reporting segment information below has been prepared based on the Company's two historical reporting segments, which were utilized in managing operations for the year ended December 31, 2022.
Global Spine
The Global Spine reporting segment offers three primary product categories: Bone Growth Therapies, Spinal Implants, and Biologics.
The Bone Growth Therapies product category manufactures, distributes, and provides support services of market leading bone growth stimulator devices that enhance bone fusion. These Class III medical devices are indicated as an adjunctive, noninvasive treatment to improve fusion success rates in the cervical and lumbar spine as well as a therapeutic treatment for non-spine fractures that have not healed (non-unions). This product category uses distributors and sales representatives to sell its devices to hospitals, healthcare providers, and patients, primarily in the U.S.
The Spinal Implants product category designs, develops, and markets a broad portfolio of motion preservation and fixation implant products used in surgical procedures of the spine. Spinal Implants distributes its products through a global network of distributors and sales representatives to sell spine products to hospitals and healthcare providers.
The Biologics product category provides a portfolio of regenerative products and tissue forms that allow physicians to successfully treat a variety of spinal and orthopedic conditions. This product category specializes in the marketing of the Company’s regeneration tissue forms and distributes its tissues to hospitals and healthcare providers, primarily in the U.S., through a network of independent distributors and sales representatives. The partnership with MTF allows the Company to exclusively market the Virtuos Lyograph, Trinity Evolution, FiberFuse Advanced, FiberFuse Strip, and certain other tissue forms for musculoskeletal defects to enhance bony fusion.
Global Orthopedics
The Global Orthopedics reporting segment offers products and solutions that allow physicians to successfully treat a variety of orthopedic conditions unrelated to the spine. This reporting segment specializes in the design, development, and marketing of the Company’s orthopedic products used in fracture repair, deformity correction, and bone reconstruction procedures. Global Orthopedics distributes its products through a global network of distributors and sales representatives to sell orthopedic products to hospitals, and healthcare providers.
Corporate
Corporate activities are comprised of the operating expenses and activities of the Company not necessarily identifiable within the two reporting segments.
The table below presents net sales by major product category by reporting segment:
F-27
| | | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | |
| | 2022 | | | 2021 | | | 2020 | |
(U.S. Dollars, in thousands) | | Net Sales | | | Percent of
Total Net
Sales | | | Net Sales | | | Percent of
Total Net
Sales | | | Net Sales | | | Percent of
Total Net
Sales | |
Bone Growth Therapies | | $ | 187,247 | | | | 40.7 | % | | $ | 187,448 | | | | 40.4 | % | | $ | 171,396 | | | | 42.2 | % |
Spinal Implants | | | 109,546 | | | | 23.8 | % | | | 115,094 | | | | 24.8 | % | | | 94,857 | | | | 23.3 | % |
Biologics | | | 56,381 | | | | 12.2 | % | | | 56,421 | | | | 12.1 | % | | | 55,482 | | | | 13.6 | % |
Global Spine | | | 353,174 | | | | 76.7 | % | | | 358,963 | | | | 77.3 | % | | | 321,735 | | | | 79.1 | % |
Global Orthopedics | | | 107,539 | | | | 23.3 | % | | | 105,516 | | | | 22.7 | % | | | 84,827 | | | | 20.9 | % |
Net sales | | $ | 460,713 | | | | 100.0 | % | | $ | 464,479 | | | | 100.0 | % | | $ | 406,562 | | | | 100.0 | % |
The following table presents EBITDA, the primary metric used in managing the Company, by reporting segment:
| | | | | | | | | | | | |
| | Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Global Spine | | $ | 60,649 | | | $ | 58,014 | | | $ | 63,036 | |
Global Orthopedics | | | (4,037 | ) | | | 3,374 | | | | (4,993 | ) |
Corporate | | | (44,011 | ) | | | (31,691 | ) | | | (25,382 | ) |
Total EBITDA | | | 12,601 | | | | 29,697 | | | | 32,661 | |
Depreciation and amortization | | | (29,019 | ) | | | (29,599 | ) | | | (30,546 | ) |
Goodwill impairment | | | — | | | | (11,756 | ) | | | — | |
Interest expense, net | | | (1,288 | ) | | | (1,837 | ) | | | (2,483 | ) |
Loss before income taxes | | $ | (17,706 | ) | | $ | (13,495 | ) | | $ | (368 | ) |
The following table presents depreciation and amortization by reporting segment:
| | | | | | | | | | | | |
| | Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Global Spine | | $ | 18,213 | | | $ | 17,548 | | | $ | 18,362 | |
Global Orthopedics | | | 6,696 | | | | 8,233 | | | | 7,896 | |
Corporate | | | 4,110 | | | | 3,818 | | | | 4,288 | |
Total | | $ | 29,019 | | | $ | 29,599 | | | $ | 30,546 | |
Geographical information
The following data includes net sales by geographic destination:
| | | | | | | | | | | | |
| | Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
U.S. | | $ | 358,843 | | | $ | 361,945 | | | $ | 327,280 | |
Italy | | | 19,098 | | | | 20,187 | | | | 18,733 | |
Germany | | | 11,569 | | | | 13,716 | | | | 11,940 | |
United Kingdom | | | 10,171 | | | | 10,552 | | | | 7,147 | |
France | | | 10,377 | | | | 10,475 | | | | 8,354 | |
Brazil | | | 5,668 | | | | 5,108 | | | | 2,347 | |
Others | | | 44,987 | | | | 42,496 | | | | 30,761 | |
Net sales | | $ | 460,713 | | | $ | 464,479 | | | $ | 406,562 | |
F-28
The table below presents net sales by geographic destination for each reporting segment and for the consolidated Company:
| | | | | | | | | | | | |
| | Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Global Spine | | | | | | | | | |
U.S. | | $ | 332,846 | | | $ | 337,455 | | | $ | 304,595 | |
International | | | 20,328 | | | | 21,508 | | | | 17,140 | |
Total Global Spine | | | 353,174 | | | | 358,963 | | | | 321,735 | |
| | | | | | | | | |
Global Orthopedics | | | | | | | | | |
U.S. | | $ | 25,997 | | | | 24,490 | | | | 22,685 | |
International | | | 81,542 | | | | 81,026 | | | | 62,142 | |
Total Global Orthopedics | | | 107,539 | | | | 105,516 | | | | 84,827 | |
| | | | | | | | | |
Consolidated | | | | | | | | | |
U.S. | | | 358,843 | | | | 361,945 | | | | 327,280 | |
International | | | 101,870 | | | | 102,534 | | | | 79,282 | |
Net sales | | $ | 460,713 | | | $ | 464,479 | | | $ | 406,562 | |
The following data includes property, plant, and equipment by geographic area:
| | | | | | | | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
U.S. | | $ | 44,802 | | | $ | 45,090 | |
Italy | | | 8,535 | | | | 9,412 | |
Germany | | | 3,115 | | | | 2,544 | |
United Kingdom | | | 1,149 | | | | 1,193 | |
Brazil | | | 85 | | | | 91 | |
Others | | | 543 | | | | 922 | |
Total | | $ | 58,229 | | | $ | 59,252 | |
17. Acquisition-related amortization and remeasurement
Acquisition-related amortization and remeasurement consists of (i) amortization related to intangible assets acquired through business combinations or asset acquisitions, (ii) the remeasurement of any related contingent consideration arrangement, (iii) recognized costs associated with acquired IPR&D assets, which are recognized immediately upon acquisition, and (iv) impairments of goodwill related to previously recognized business combinations. Components of acquisition-related amortization and remeasurement for the years ended December 31, 2022, 2021, and 2020, respectively, are as follows:
| | | | | | | | | | | | |
| | Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Changes in fair value of contingent consideration | | $ | (17,200 | ) | | $ | (3,575 | ) | | $ | (7,300 | ) |
Amortization of acquired intangibles | | | 8,196 | | | | 7,907 | | | | 6,801 | |
Acquired IPR&D | | | 1,600 | | | | 1,500 | | | | — | |
Impairment of Global Orthopedics goodwill | | | — | | | | 11,756 | | | | — | |
Total | | $ | (7,404 | ) | | $ | 17,588 | | | $ | (499 | ) |
CGBio Co. Ltd. License and Distribution Agreement
On July 30, 2022, the Company entered into an exclusive License and Distribution Agreement (the "License Agreement") with CGBio Co., Ltd. (“CGBio”), a developer of innovative, synthetic bone grafts. The Agreement grants the Company the exclusive right to conduct pre-clinical and clinical studies, commercialize, promote, market, and sell the Novosis recombinant human bone morphogenetic protein-2 (rhBMP-2) bone growth materials and other future tissue regenerative solutions in the U.S. and Canada. As consideration, the Company agreed to pay CGBio an upfront payment of $1.4 million with additional payments contingent upon the achievement of specified development milestones. The Company accounted for this transaction as an asset acquisition. As the
F-29
transaction was classified as an asset acquisition, the value of the consideration associated with the contingent milestones will be recognized at the time that applicable contingencies are resolved and consideration is paid or becomes payable. The $1.4 million upfront payment was paid in the third quarter of 2022 and was recognized as acquired IPR&D costs, which was then immediately expensed.
Legion Innovations, LLC Asset Acquisition
On December 29, 2022, the Company entered into a technology assignment and royalty agreement with Legion Innovations, LLC, a U.S.-based medical device technology company, whereby the Company acquired intellectual property rights to certain assets. As consideration, the Company agreed to pay $0.2 million in January 2023, with additional payments contingent upon reaching future commercialization and revenue-based milestones. The Company accounted for this transaction as an asset acquisition. As the transaction was classified as an asset acquisition, the value of the consideration associated with the contingent milestones will be recognized at the time that applicable contingencies are resolved and consideration is paid or becomes payable. The $0.2 million initial payment was accrued as of December 31, 2022, and was recognized as acquired IPR&D costs, which was then immediately expensed.
IGEA S.p.A Asset Acquisition
In April 2021, the Company entered into an Exclusive License and Distribution Agreement (the “License Agreement”) with IGEA S.p.A (“IGEA”), an Italian manufacturer and distributor of bone and cartilage stimulation systems. As consideration for the License Agreement, the Company agreed to pay up to $4.0 million, with certain payments contingent upon reaching an FDA milestone. Of this amount, $0.5 million was paid in 2021, which was recognized as acquired IPR&D costs within acquisition-related amortization and remeasurement. The Company accounted for this transaction as an asset acquisition. As the transaction was classified as an asset acquisition, the value of the consideration associated with the contingent milestones will be recognized at the time that applicable contingencies are resolved and consideration is paid or becomes payable. The License Agreement also includes certain minimum purchase requirements.
In May 2022, the Company achieved FDA approval pertaining to the acquired technology, triggering a contingent consideration milestone obligation of $3.5 million. Of this amount, $1.5 million was paid in 2022, $1.0 million was accrued within other current liabilities, and $1.0 million was accrued within other long-term liabilities as of December 31, 2022.
Related Party Asset Acquisition
In February 2021, the Company entered into a technology assignment and royalty agreement with a medical device technology company partially owned and controlled by the wife of our Executive Chairman, and former President and Chief Executive Officer, Jon Serbousek, whereby the Company acquired the intellectual property rights to certain assets for consideration of up to $10.0 million.
Consideration was comprised of $1.0 million due at signing, which was recognized immediately as acquired IPR&D expense within acquisition-related amortization and remeasurement, and $9.0 million in contingent consideration. The contingent consideration is dependent upon multiple milestones, such as receipt of 510(k) clearance and the attainment of certain net sales targets. The Company accounted for this transaction as an asset acquisition. As the transaction was classified as an asset acquisition, the value of the consideration associated with the contingent milestones will be recognized at the time that applicable contingencies are resolved and consideration is paid or becomes payable. In addition, the Company is obligated to pay a royalty of 2% to 4% on net sales, commencing upon commercialization of the assets.
The transaction was approved by the Company’s Audit and Finance Committee, with the Audit and Finance Committee directly supervising the negotiations of the transaction. Mr. Serbousek was excluded from such discussions and did not participate in the negotiation or evaluation of the transaction. Mr. Serbousek also continues to be excluded from the oversight of the Company’s development and commercialization activities in relation to the acquired technology and all other matters relating to the relationship between the Company and the counterparty
F-30
18. Share-based compensation
At December 31, 2022, and 2021, the Company had stock option and award plans, and a stock purchase plan.
2012 Long Term Incentive Plan
The Board of Directors adopted the Amended and Restated 2012 Long-Term Incentive Plan (the “2012 LTIP”) on April 23, 2018, which was subsequently approved by shareholder ratification. The 2012 LTIP provides for the grant of options to purchase shares of the Company’s common stock, stock awards (including restricted stock, unrestricted stock, and stock units), stock appreciation rights, performance-based awards and other equity-based awards. All of the Company’s employees and the employees of the Company’s subsidiaries and affiliates are eligible and may receive awards under the 2012 LTIP. In addition, the Company’s non-employee directors, consultants, and advisors who perform services for the Company and its subsidiaries and affiliates may receive awards under the 2012 LTIP. Awards granted under the 2012 LTIP expire no later than ten years after the date of grant. At December 31, 2022, the Company reserves a total of 8,375,000 shares of common stock for issuance pursuant to the 2012 LTIP, subject to certain adjustments set forth in the 2012 LTIP. At December 31, 2022, there were 1,098,680 options outstanding under the 2012 LTIP, of which 852,490 were exercisable. In addition, there were 1,359,693 restricted stock units outstanding, some of which contain performance-based or market-based vesting conditions, under the 2012 LTIP as of December 31, 2022.
Inducement Plans
In 2013, the Company granted options to acquire up to 150,000 shares of common stock to a former Chief Executive Officer as an inducement to accept employment with the Company. As of December 31, 2022, there were 150,000 options outstanding under this inducement, all of which were exercisable.
In August 2019, the Company appointed a new President of Global Spine, who was then subsequently promoted to President and Chief Executive Officer. As an inducement to accept employment with the Company, the individual was awarded a grant of stock options to acquire up to 50,711 shares of common stock and an award of 14,743 restricted stock units. As of December 31, 2022, there were 50,711 options outstanding under this inducement, 38,033 of which were exercisable, and 3,686 unvested restricted stock units outstanding.
Stock Purchase Plan
The Second Amended and Restated Stock Purchase Plan, as Amended (the “Stock Purchase Plan”) provides for the issuance of shares of the Company’s common stock to eligible employees and directors of the Company and its subsidiaries that elect to participate in the plan and acquire shares of common stock through payroll deductions (including executive officers).
During each purchase period, eligible employees may designate between 1% and 25% of their compensation to be deducted for the purchase of common stock under the plan (or such other percentage in order to comply with regulations applicable to employees domiciled in or resident of a member state of the European Union). For eligible directors, the designated percentage will be applied to an amount equal to his or her director compensation paid in cash for the current plan period. The purchase price of the shares under the plan is equal to 85% of the fair market value on the first day of the plan period or, if lower, on the last day of the plan period.
Due to the compensatory nature of such plan, the Company records the related share-based compensation expense in the consolidated statement of operations. Compensation expense is estimated using the Black-Scholes valuation model, with such value recognized as expense over the plan period. As of December 31, 2022, the aggregate number of shares reserved for issuance under the Stock Purchase Plan is 2,850,000. As of December 31, 2022, a total of 2,395,673 shares had been issued pursuant to the Stock Purchase Plan.
F-31
Share-Based Compensation Expense
Share-based compensation expense is recorded in the same line of the consolidated statements of operations as the employee’s cash compensation. The following tables present the detail of share-based compensation expense by line item in the consolidated statements of income as well as by award type, for the years ended December 31, 2022, 2021, and 2020:
| | | | | | | | | | | | |
| | Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Cost of sales | | $ | 826 | | | $ | 779 | | | $ | 705 | |
Sales and marketing | | | 3,865 | | | | 3,385 | | | | 3,620 | |
General and administrative | | | 12,917 | | | | 10,289 | | | | 10,624 | |
Research and development | | | 835 | | | | 979 | | | | 1,258 | |
Total | | $ | 18,443 | | | $ | 15,432 | | | $ | 16,207 | |
| | | | | | | | | | | | |
| | Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
Stock options | | $ | 1,114 | | | $ | 1,893 | | | $ | 2,571 | |
Time-based restricted stock awards and stock units | | | 9,452 | | | | 7,437 | | | | 8,485 | |
Performance-based / Market-based restricted stock units | | | 6,425 | | | | 4,414 | | | | 3,509 | |
Stock purchase plan | | | 1,452 | | | | 1,688 | | | | 1,642 | |
Total | | $ | 18,443 | | | $ | 15,432 | | | $ | 16,207 | |
The income tax benefit related to this expense was $3.3 million, $3.1 million, and $3.2 million for the years ended December 31, 2022, 2021, and 2020, respectively.
Stock Options
The fair value of time-based stock options is determined using the Black-Scholes valuation model, with such value recognized as expense over the service period, which is typically four years, net of actual forfeitures. A summary of the Company’s assumptions used in determining the fair value of the stock options granted during each of the years ended December 31, 2022, 2021, and 2020, is shown in the following table. The Company did not grant any time-based stock options in 2022.
| | | | | | | | | | | | |
| | Year Ended December 31, | |
| | 2022 | | | 2021 | | | 2020 | |
Assumptions: | | | | | | | | | |
Expected term (in years) | | | — | | | 6.0 | | | 5.5 | |
Expected volatility | | | — | | | 34.4% – 34.8% | | | 30.2% – 35.1% | |
Risk free interest rate | | | — | | | 0.83% – 1.25% | | | 0.28% – 1.65% | |
Dividend yield | | | — | | | | — | | | | — | |
Weighted average grant date fair value | | | — | | | $ | 12.33 | | | $ | 8.74 | |
The expected term of the options granted is estimated based on a number of factors, including the vesting and expiration terms of the award, historical employee exercise behavior for both options that are currently outstanding and options that have been exercised or are expired, and an employee’s average length of service. Expected volatility is based on the historical volatility of the Company’s common stock. The risk-free interest rate is determined based upon a constant U.S. Treasury security rate with a contractual life that approximates the expected term of the option.
F-32
Summaries of the status of the Company’s stock option plans as of December 31, 2022, and 2021, and changes during the year ended December 31, 2022, are presented below:
| | | | | | | | | | | | |
| | Options | | | Weighted
Average
Exercise
Price | | | Weighted
Average
Remaining
Contractual
Term | |
Outstanding at December 31, 2021 | | | 1,397,054 | | | $ | 39.20 | | | | |
Granted | | | — | | | $ | - | | | | |
Exercised | | | (575 | ) | | $ | 21.78 | | | | |
Forfeited or expired | | | (97,088 | ) | | $ | 38.14 | | | | |
Outstanding at December 31, 2022 | | | 1,299,391 | | | $ | 39.29 | | | | 3.77 | |
Vested and expected to vest at December 31, 2022 | | | 1,299,391 | | | $ | 39.29 | | | | 3.77 | |
Exercisable at December 31, 2022 | | | 1,040,523 | | | $ | 40.70 | | | | 3.13 | |
As of December 31, 2022, the unamortized compensation expense relating to options granted and expected to be recognized was $0.8 million. This amount is expected to be recognized through December 2025 over a weighted average period of approximately 1.0 years. The total intrinsic value of options exercised was $0.0 million, $0.6 million, and $0.9 million for the years ended December 31, 2022, 2021, and 2020, respectively. For the year ended December 31, 2022, we received $0.0 million in cash from stock option exercises, with the tax benefit realized for the tax deductions from these exercises of $0.0 million. The aggregate intrinsic value of options outstanding and options exercisable as of December 31, 2022, is calculated as the difference between the exercise price of the underlying options and the market price of the Company’s common stock for options that had exercise prices lower than $20.53, the closing price of the Company’s stock on December 31, 2022. The aggregate intrinsic value of options outstanding was $0.0 million as of December 31, 2022. The aggregate intrinsic value of options exercisable was also $0.0 million as of that date.
Time-based Restricted Stock Awards and Stock Units
Compensation expense for time-based restricted stock awards and stock units, which represents the fair value of the stock measured at the market price at the date of grant, is recognized on a straight-line basis over the vesting period, which is typically four years, net of actual forfeitures.
Since 2017, the annual grant to non-employee directors has been made in the form of one-year vesting restricted stock units with deferred delivery (“DSUs”), whereby shares are not settled until after the director ceases service as a director. As of December 31, 2022, there were 86,542 DSUs outstanding that are vested but not settled.
The aggregate fair value of time-based restricted stock awards and stock units that vested during the years ended December 31, 2022, 2021, and 2020, was $5.2 million, $9.0 million, and $6.5 million, respectively. Unamortized compensation expense related to time-based restricted stock awards and stock units amounted to $18.6 million at December 31, 2022. This amount is expected to be recognized through October 2026 over a weighted average period of approximately 2.5 years. The aggregate intrinsic value of time-based restricted stock awards and stock units outstanding was $17.4 million as of December 31, 2022.
Performance-based and Market-based Restricted Stock Units
Certain of the Company's outstanding restricted stock units contain performance-based vested conditions or market-based vesting conditions.
The fair value of performance-based restricted stock units is calculated based upon the closing stock price at the date of grant. Such value is recognized as expense over the requisite service period beginning in the period in which they are deemed probable to vest, net of actual forfeitures. Vesting probability is assessed based upon forecasted earnings and financial results.
The fair value of market-based restricted stock units is determined at the date of the grant using the Monte Carlo valuation methodology, with any discounts for post-vesting restrictions estimated using the Chaffe Model. The Monte Carlo methodology incorporates into the valuation the possibility that the market condition may not be satisfied. Such value is recognized on a straight-line basis over the vesting period, net of actual forfeitures.
F-33
The fair value of performance-based and/or market-based restricted stock units that vested and settled during the years ended December 31, 2022, 2021, and 2020, totaled $0.0 million, $0.0 million, and $1.4 million, respectively. Unamortized compensation expense for performance-based and/or market-based restricted stock units totaled $9.3 million at December 31, 2022, and is expected to be recognized over a weighted average period of approximately 1.4 years. The aggregate intrinsic value of market-based restricted stock units outstanding was $10.6 million as of December 31, 2022.
A summary of the status of our time-based and performance-based and/or market-based restricted stock units as of December 31, 2022, and 2021, and changes during the year ended December 31, 2022, are presented below:
| | | | | | | | | | | | | | | | |
| | Time-based Restricted Stock
Awards and Stock Units | | | Performance-based and/or Market-based
Restricted Stock Units | |
| | Shares | | | Weighted
Average Grant
Date Fair Value | | | Shares | | | Weighted
Average Grant
Date Fair Value | |
Outstanding at December 31, 2021 | | | 559,368 | | | $ | 40.03 | | | | 323,081 | | | $ | 48.56 | |
Granted | | | 500,222 | | | $ | 29.81 | | | | 255,327 | | | $ | 33.57 | |
Vested and settled | | | (152,735 | ) | | $ | 41.03 | | | | — | | | $ | — | |
Cancelled | | | (59,708 | ) | | $ | 34.80 | | | | (62,176 | ) | | $ | 55.70 | |
Outstanding at December 31, 2022 | | | 847,147 | | | $ | 34.18 | | | | 516,232 | | | $ | 40.29 | |
19. Defined contribution plans and deferred compensation
Defined Contribution Plans
Orthofix US LLC sponsors a defined contribution plan (the “401(k) Plan”) covering substantially all full-time U.S. employees. The 401(k) Plan allows participants to contribute up to 80% of their pre-tax compensation, subject to certain limitations, with the Company matching 100% of the first 2% of the employee’s base compensation and 50% of the next 4% of the employee’s base compensation if contributed to the 401(k) Plan. During the years ended December 31, 2022, 2021, and 2020, expenses incurred relating to the 401(k) Plan, including matching contributions, were approximately $3.3 million, $2.8 million, and $1.1 million, respectively.
In April 2020, as a precautionary measure to increase the Company’s cash position and preserve financial flexibility in response to the initial uncertainty of the COVID-19 pandemic, the Company temporarily suspended the 401(k) match program through the remainder of fiscal year 2020. The 401(k) match program was reinstated in January 2021.
The Company also operates defined contribution plans for its international employees meeting minimum service requirements. The Company’s expenses for such contributions during each of the years ended December 31, 2022, 2021, and 2020, were $1.1 million, $1.2 million, and $1.1 million, respectively.
Deferred Compensation Plans
Under Italian Law, our Italian subsidiary accrues deferred compensation on behalf of its employees, which is paid on termination of employment. The accrual for deferred compensation is based on a percentage of the employee’s current annual remuneration plus an annual charge. Deferred compensation is also accrued for the leaving indemnity payable to agents in case of dismissal, which is regulated by a national contract and is equal to approximately 4% of total commissions earned from the Company. The Company’s relations with its Italian employees, who represent 21% of total employees at December 31, 2022, are governed by the provisions of a National Collective Labor Agreement setting forth mandatory minimum standards for labor relations in the metal mechanic workers industry. The Company is not a party to any other collective bargaining agreement. The balance in other long-term liabilities as of December 31, 2022, and 2021 was $1.5 million and $1.3 million, respectively, and represents the amount that would be payable if all the employees and agents had terminated employment at that date.
F-34
20. Income taxes
Income (loss) before provision for income taxes consisted of the following:
| | | | | | | | | | | | |
| | Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
U.S. | | $ | (22,318 | ) | | $ | (5,987 | ) | | $ | 5,556 | |
Non-U.S. | | | 4,612 | | | | (7,508 | ) | | | (5,924 | ) |
Income (loss) before income taxes | | $ | (17,706 | ) | | $ | (13,495 | ) | | $ | (368 | ) |
The provision for income taxes consists of the following:
| | | | | | | | | | | | |
| | Year Ended December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | | | 2020 | |
U.S. | | | | | | | | | |
Current | | $ | 1,151 | | | $ | (607 | ) | | $ | (15,054 | ) |
Deferred | | | 67 | | | | 24,292 | | | | (29 | ) |
| | | 1,218 | | | | 23,685 | | | | (15,083 | ) |
Non-U.S. | | | | | | | | | |
Current | | | 578 | | | | 1,009 | | | | 1,382 | |
Deferred | | | 247 | | | | 190 | | | | 10,816 | |
| | | 825 | | | | 1,199 | | | | 12,198 | |
Income tax expense (benefit) | | $ | 2,043 | | | $ | 24,884 | | | $ | (2,885 | ) |
The differences between the income tax provision at the U.S. federal statutory tax rate and the Company’s effective tax rate for the years ended December 31, 2022, 2021, and 2020, consist of the following:
| | | | | | | | | | | | | | | | | | | | | | | | |
| | 2022 | | | 2021 | | | 2020 | |
(U.S. Dollars, in thousands, except percentages) | | Amount | | | Percent | | | Amount | | | Percent | | | Amount | | | Percent | |
Statutory U.S. federal income tax rate | | $ | (3,718 | ) | | | 21.0 | % | | $ | (2,834 | ) | | | 21.0 | % | | $ | (77 | ) | | | 21.0 | % |
State taxes, net of U.S. federal benefit | | | (1,312 | ) | | | 7.4 | | | | (24 | ) | | | 0.2 | | | | 1,151 | | | | (312.8 | ) |
Foreign rate differential, including withholding taxes | | | 475 | | | | (2.7 | ) | | | 480 | | | | (3.6 | ) | | | (147 | ) | | | 39.9 | |
Valuation allowances, net | | | 7,638 | | | | (43.1 | ) | | | 27,819 | | | | (206.1 | ) | | | 14,514 | | | | (3,944.0 | ) |
Foreign income inclusions, net | | | 1,018 | | | | (5.7 | ) | | | — | | | | — | | | | — | | | | — | |
Research credits | | | (750 | ) | | | 4.2 | | | | (537 | ) | | | 4.0 | | | | (982 | ) | | | 266.8 | |
Unrecognized tax benefits, net of settlements | | | (599 | ) | | | 3.4 | | | | (1,363 | ) | | | 10.1 | | | | (17,321 | ) | | | 4,706.8 | |
Equity compensation | | | 1,441 | | | | (8.1 | ) | | | 1,091 | | | | (8.1 | ) | | | 1,657 | | | | (450.3 | ) |
Executive compensation | | | 697 | | | | (3.9 | ) | | | 456 | | | | (3.4 | ) | | | 375 | | | | (101.9 | ) |
Contingent consideration | | | (3,316 | ) | | | 18.7 | | | | (640 | ) | | | 4.7 | | | | (1,460 | ) | | | 396.7 | |
Other, net | | | 469 | | | | (2.6 | ) | | | 436 | | | | (3.2 | ) | | | (595 | ) | | | 161.8 | |
Income tax expense (benefit) /effective rate | | $ | 2,043 | | | | (11.5 | )% | | $ | 24,884 | | | | (184.4 | )% | | $ | (2,885 | ) | | | 784.0 | % |
The Company paid (received or was refunded) cash relating to taxes totaling ($0.9) million, $4.8 million, and $0.5 million for the years ended December 31, 2022, 2021, and 2020, respectively.
F-35
The Company’s deferred tax assets and liabilities are as follows:
| | | | | | | | |
| | December 31, | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
Intangible assets and goodwill | | $ | 5,807 | | | $ | 5,245 | |
Inventories and related reserves | | | 17,819 | | | | 17,097 | |
Deferred revenue and cost of goods sold | | | 3,642 | | | | 3,888 | |
Other accruals and reserves | | | 2,756 | | | | 3,082 | |
Accrued compensation | | | 8,795 | | | | 7,784 | |
Provision for expected credit losses | | | 1,253 | | | | 1,217 | |
Net operating loss and tax credit carryforwards | | | 40,676 | | | | 42,546 | |
R&D capitalization | | | 4,353 | | | | — | |
Lease liabilities | | | 6,440 | | | | 5,691 | |
Other, net | | | 3,767 | | | | 852 | |
Total deferred tax assets | | | 95,308 | | | | 87,402 | |
Valuation allowance | | | (83,797 | ) | | | (76,725 | ) |
Deferred tax asset, net of valuation allowance | | $ | 11,511 | | | $ | 10,677 | |
Withholding taxes | | | (10 | ) | | | (10 | ) |
Property, plant, and equipment | | | (5,516 | ) | | | (4,809 | ) |
Right-of-use lease assets | | | (5,771 | ) | | | (5,165 | ) |
Deferred tax liability | | | (11,297 | ) | | | (9,984 | ) |
Net deferred tax assets | | $ | 214 | | | $ | 693 | |
| | | | | | |
Reported as: | | | | | | |
Deferred income tax assets (classified within other long-term assets) | | | 1,470 | | | | 1,771 | |
Deferred income tax liabilities (classified within other long-term liabilities) | | | (1,256 | ) | | | (1,078 | ) |
Net deferred tax assets | | $ | 214 | | | $ | 693 | |
The Company historically presented deferred income tax assets as a separate and discrete line item on its consolidated balance sheet; however, as the significance of the asset has decreased as a result of the recognition of valuation allowances, the Company has reclassified this balance to be included within other long-term assets. As such, the prior year balance has been reclassified to conform to current period presentation.
The Company accounts for income taxes using the asset and liability method, under which deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the financial reporting and income tax basis of assets and liabilities, and for operating losses and credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates in effect for the years in which those items are expected to be realized. Tax law and rate changes are recorded in the period such changes are enacted. The Company establishes a valuation allowance when it is more likely than not that certain deferred tax assets will not be realized in the foreseeable future.
The valuation allowance is primarily attributable to net operating loss carryforwards and temporary differences in domestic and certain foreign jurisdictions. The net increase in the valuation allowance of $7.1 million during the year principally relates to recognizing a full valuation allowance against the net deferred tax asset within the Company’s U.S. operations as well as an increase in valuation allowance against deferred tax assets within the Company’s Italian manufacturing subsidiary. The Company considered many factors when assessing the likelihood of future realization of these deferred tax assets, including recent cumulative losses experienced by the subsidiary, expectations of future taxable income or loss, the carryforward periods available to the Company for tax reporting purposes, and other relevant factors. That increase was partially offset by a decrease of valuation allowances on net operating loss carryforwards in other foreign jurisdictions due to expiration, statutory rate changes, and changes regarding the realizability of net deferred tax assets. It is reasonably possible that the valuation allowance will increase in 2023 due to further losses in certain jurisdictions, offset by decreases related to the expiration of foreign net operating losses.
The Company has federal net operating loss carryforwards of $13.3 million and research and development credits of $1.7 million, including amounts from the acquisition of Spinal Kinetics. These carryforwards are subject to limitation under the provisions of Section 382 and will begin to expire in 2026. The Company has state net operating loss carryforwards of approximately $31.1 million,
F-36
of which $20.6 million relates to Spinal Kinetics and begins to expire in 2027. Additionally, the Company has net operating loss carryforwards in various foreign jurisdictions of approximately $120.4 million, the majority of which do not have an expiration, which mainly relate to the Company’s Italy, Netherlands, and Brazil operations.
Unremitted foreign earnings were $27.0 million as of December 31, 2022. The Company’s investment in foreign subsidiaries continues to be indefinite in nature; however, the Company may periodically repatriate a portion of these earnings to the extent that it does not incur significant additional tax liability. Quantification of the deferred tax liability, if any, associated with indefinitely reinvested earnings of foreign subsidiaries is not practicable.
The Company records a benefit for uncertain tax positions when the weight of available evidence indicates that it is more likely than not, based on an evaluation of the technical merits, that the tax position will be sustained on audit. The tax benefit is measured as the largest amount that is more than 50% likely to be realized upon settlement. The Company re-evaluates income tax positions periodically to consider changes in facts or circumstances such as changes in or interpretations of tax law, effectively settled issues under audit, and new audit activity. The Company includes interest and any applicable penalties related to income tax issues as part of income tax expense in its consolidated financial statements.
The Company’s unrecognized tax benefit was $1.7 million and $3.5 million for the years ended December 31, 2022, and 2021, respectively. The Company recorded net interest and penalties expense (benefit) on unrecognized tax benefits of $0.1 million, $(0.4) million, and $(5.4) million for the years ended December 31, 2022, 2021, and 2020, respectively, and had approximately $0.9 million and $0.8 million accrued for payment of interest and penalties as of December 31, 2022, and 2021, respectively. The entire amount of unrecognized tax benefits, including interest, would favorably impact the Company’s effective tax rate if recognized. The Company believes it is reasonably possible that, in the next 12 months, no unrecognized tax benefits, exclusive of interest and penalties, will be resolved by the closure of an audit or statute release.
A reconciliation of the gross unrecognized tax benefits (excluding interest and penalties) for the years ended December 31, 2022, and 2021, is shown below:
| | | | | | | | |
(U.S. Dollars, in thousands) | | 2022 | | | 2021 | |
Balance as of January 1, | | $ | 3,462 | | | $ | 4,629 | |
Additions for current year tax positions | | | 46 | | | | 45 | |
Increases for prior year tax positions | | | 16 | | | | 110 | |
Settlements of prior year tax positions | | | (144 | ) | | | — | |
Expiration of statutes | | | (1,637 | ) | | | (1,322 | ) |
Balance as of December 31, | | $ | 1,743 | | | $ | 3,462 | |
The Company and its subsidiaries file income tax returns in the U.S. federal jurisdiction and in certain state and foreign jurisdictions, including Italy, as well as other jurisdictions where the Company maintains operations. The statute of limitations with respect to federal and state tax filings is closed for years prior to 2019. The statute of limitations with respect to the major foreign tax filing jurisdictions is closed for years prior to 2018. The Company cannot reasonably determine if any state and local or foreign examinations will have a material impact on its financial statements and cannot predict the timing regarding the resolution of these tax examinations.
21. Earnings per share (EPS)
The Company uses the two-class method of computing basic EPS due to the existence of non-vested restricted stock awards in certain periods with nonforfeitable rights to dividends or dividend equivalents (referred to as participating securities). Basic EPS is computed using the weighted average number of common shares outstanding during each of the respective years. Diluted EPS is computed using the weighted average number of common and common equivalent shares outstanding during each of the respective years using the more dilutive of either the treasury stock method or two-class method. The difference between basic and diluted shares, if any, largely results from common equivalent shares, which represents the dilutive effect of the assumed exercise of certain outstanding share options, the assumed vesting of restricted stock granted to employees and directors, or the satisfaction of certain necessary conditions for contingently issuable shares (see Note 18).
F-37
For each of the three years ended December 31, 2022, 2021, and 2020, no significant adjustments were made to net income for purposes of calculating basic and diluted EPS. The following is a reconciliation of the weighted average shares used in the diluted EPS computations:
| | | | | | | | | | | | |
| | Year Ended December 31, | |
| | 2022 | | | 2021 | | | 2020 | |
Weighted average common shares-basic | | | 20,053,548 | | | | 19,690,593 | | | | 19,267,920 | |
Effect of diluted securities: | | | | | | | | | |
Unexercised stock options and employee stock purchase plan | | | — | | | | — | | | | 51,951 | |
Unvested time-based restricted stock units | | | — | | | | — | | | | 71,847 | |
Weighted average common shares-diluted | | | 20,053,548 | | | | 19,690,593 | | | | 19,391,718 | |
There were 2,313,226; 1,711,323; and 1,499,630 weighted average outstanding options, restricted stock, and market-based units not included in the diluted earnings per share computation for the years ended December 31, 2022, 2021, and 2020, respectively, because inclusion of these awards was anti-dilutive or, for market-based units, all necessary conditions had not been satisfied by the end of the respective period.
22. Subsequent Events
Merger with SeaSpine Holdings Corporation
On October 10, 2022, the Company and Orca Merger Sub Inc., a wholly-owned subsidiary of Orthofix (“Merger Sub”), entered into an Agreement and Plan of Merger (the “Merger Agreement”) with SeaSpine, a global medical technology company focused on surgical solutions for the treatment of spinal disorders. On January 5, 2023, the transaction was completed and Merger Sub was merged with and into SeaSpine (the “Merger”), with SeaSpine continuing as the surviving company and a wholly-owned subsidiary of Orthofix following the transaction. For the year ended December 31, 2022, the Company incurred approximately $9.3 million in acquisition-related costs, substantially all of which was classified as general and administrative expenses.
As a result of the Merger, each share of common stock of SeaSpine issued and outstanding immediately prior to the closing date was converted into the right to receive 0.4163 shares of common stock of Orthofix. In addition, the Company assumed SeaSpine’s existing equity incentive plans in connection with the Merger, and outstanding SeaSpine equity awards were converted into Orthofix equity awards (on the same vesting schedule and other terms and conditions as existed prior to such conversion). The conversion of such equity awards occurred at the same exchange ratio as applied to SeaSpine common stock in the Merger, and the exercise price of converted SeaSpine stock options was also correspondingly adjusted.
The Merger is being accounted for as an acquisition of SeaSpine by Orthofix under the acquisition method of accounting for business combinations in accordance with U.S. GAAP. Thus, Orthofix is treated as the acquirer for accounting purposes. In identifying the acquirer, Orthofix and SeaSpine considered the structure of the transaction and other actions contemplated by the Merger Agreement, relative outstanding share ownership, and market values, the composition of the combined company’s board of directors, and the relative size of Orthofix and SeaSpine.
The total estimated fair value of consideration associated with the Merger as of the acquisition date was comprised of:
| | | | |
(U.S. Dollars, in thousands, except shares and price per share) | | | |
Share Consideration: | | | |
Orthofix common shares to be issued in exchange for SeaSpine common shares | | | 16,104,854 | |
Orthofix closing price per share as of January 4, 2023 | | $ | 22.76 | |
Estimated fair value of shares issued in exchange for SeaSpine common shares | | $ | 366,546 | |
Estimated fair value of Orthofix stock options and RSUs issued in exchange for outstanding SeaSpine equity awards | | | 11,508 | |
Total estimated fair value of consideration | | $ | 378,054 | |
Pursuant to the Merger Agreement, the outstanding equity awards of SeaSpine (including stock options and restricted stock units) were exchanged for awards of Orthofix. The Company issued 1,889,812 options to purchase shares of Orthofix common stock and 490,338 shares of time-based vesting restricted stock in the conversion of such awards. The estimated fair value of the portion of the SeaSpine awards for which the required service period had been completed at the time of the acquisition was treated as
F-38
purchase consideration. The remaining estimated fair value will be recorded as compensation expense over the remainder of the service period associated with the awards.
As of the date the consolidated financial statements were available to be issued, the Company was in the process of determining a preliminary allocation of the total estimated fair value of consideration to the fair value of the net assets acquired and residual goodwill; however, such allocation has not yet been finalized. A preliminary allocation of the purchase price to assets acquired and liabilities assumed will be reported in the Company’s quarterly report on Form 10-Q for the quarter ended March 31, 2023; however, such allocation will be subject to completion of the Company’s valuation of the assets acquired and liabilities assumed, which the Company expects to complete within one year from the acquisition date.
In connection with the Merger, immediately after the closing of the acquisition on January 5, 2023, Orthofix repaid on behalf of SeaSpine all of the outstanding obligations in respect of principal, interest and fees under an Amended and Restated Credit Agreement, dated July 27, 2018, among SeaSpine and Project Maple Leaf Holdings ULC, as guarantors, and SeaSpine Orthopedics Corporation, SeaSpine, Inc., ISOTIS, Inc., SeaSpine Sales LLC, ISOTIS Orthobiologics, Inc., Theken Spine, LLC, SeaSpine Orthopedics Intermediate Co, Inc., 7D Surgical USA Inc. and 7D Surgical ULC, as borrowers, the lenders party thereto and Wells Fargo Bank, National Association, as administrative agent, and terminated all applicable commitments under such agreement.
In addition, on January 3, 2023, the Company borrowed $30.0 million under its $300.0 million secured revolving credit facility for working capital purposes, including to fund certain merger-related expenses. Further, an additional $15.0 million was borrowed on March 3, 2023.
F-39